1. データの読み込みと整形(前回と同様)
# 前回同様にデータを読み込んで整形します。 ただし今回は正規化やlog変換は必要ありません 。 tidyverseとhereライブラリの読み込み library (tidyverse)
library (here)前回同様にcountsとmetadata変数にデータを読み込む # データの読み込み
counts <- read_csv (here ("data" , "TCGA_GTEx_colon_counts_tibble.csv" ))
metadata <- read_csv (here ("data" , "TCGA_GTEx_colon_metadata_tibble.csv" )) Rows: 61569 Columns: 103
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (3 ): gene_id, gene_name, gene_type
dbl (100 ): TCGA- D5-5540 , TCGA- EI-6509 , TCGA- A6-6137 , TCGA- QG- A5Z2, TCGA- AA-3 ...
ℹ Use `spec ()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE ` to quiet this message. Rows: 100 Columns: 210
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (210 ): case_id, source, project.project_id, cases.consent_type, cases.da...
ℹ Use `spec ()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE ` to quiet this message. gene_typeがprotein_codingのデータを抽出し同じ遺伝子名の値を合計して統合し、matrixに変換してデータを確認 # gene_typeがprotein_codingのデータを抽出
counts_mrna <- counts %>%
filter (gene_type == "protein_coding" )
# 同じ遺伝子名の値を合計して統合し、matrixに変換
counts_mrna_matrix <- counts_mrna %>%
group_by (gene_name) %>%
summarise (across (where (is.numeric), sum), .groups = 'drop' ) %>%
column_to_rownames ("gene_name" ) %>%
as.matrix ()
# データの確認
print (counts_mrna_matrix[1: 5 , 1 : 5 ])
print (dim (counts_mrna_matrix))
head (metadata) TCGA- D5-5540 TCGA- EI-6509 TCGA- A6-6137 TCGA- QG- A5Z2 TCGA- AA-3489
A1BG 1 4 0 5 5
A1CF 786 2748 1989 646 657
A2M 2342 6609 14075 4413 38490
A2ML1 1 9 17 7 8
A3GALT2 0 1 1 0 0 # A tibble: 6 × 210
case_id source project.project_id cases.consent_type cases.days_to_consent
< chr> < chr> < chr> < chr> < chr>
1 TCGA- D5-55 … TCGA_… TCGA- COAD Informed Consent 20
2 TCGA- EI-65 … TCGA_… TCGA- READ Informed Consent 0
3 TCGA- A6-61 … TCGA_… TCGA- COAD Informed Consent 0
4 TCGA- QG- A5… TCGA_… TCGA- COAD Informed Consent 49
5 TCGA- AA-34 … TCGA_… TCGA- COAD Informed Consent 31
6 HCM- CSHL-0 … TCGA_… HCMI- CMDC '-- ' --
# ℹ 205 more variables: cases.days_to_lost_to_followup <chr>,
# cases.disease_type <chr>, cases.index_date <chr>,
# cases.lost_to_followup <chr>, cases.primary_site <chr>,
# demographic.age_at_index <chr>, demographic.age_is_obfuscated <chr>,
# demographic.cause_of_death <chr>, demographic.cause_of_death_source <chr>,
# demographic.country_of_birth <chr>,
# demographic.country_of_residence_at_enrollment <chr>, … 今回はそれぞれのサンプルが腫瘍か正常か のmetadataだけあれば十分です。 sample_typeという変数にcase_idと腫瘍かどうかの列(sample_type)を作成します。 メタデータを必要な列に絞り、tumorかnormalかのメタデータにする # メタデータを必要な列に絞る
metadata_subset <- metadata %>%
select (case_id, source)
# がんか非癌かのメタデータにする
metadata_sample_type <- metadata_subset %>%
mutate (sample_type = if_else (source == "TCGA_CRC" , "tumor" , "normal" )) %>%
select (- source)
# データの確認
print (metadata_sample_type)
print (table (metadata_sample_type$ sample_type))# A tibble: 100 × 2
case_id sample_type
< chr> < chr>
1 TCGA- D5-5540 tumor
2 TCGA- EI-6509 tumor
3 TCGA- A6-6137 tumor
4 TCGA- QG- A5Z2 tumor
5 TCGA- AA-3489 tumor
6 HCM- CSHL-0160 - C18 tumor
7 TCGA- AD- A5EK tumor
8 TCGA- AA-3867 tumor
9 TCGA- AA-3975 tumor
10 05 CO007 tumor
# ℹ 90 more rows 2. DEG解析(DEseq2パッケージ)
# DEG解析を行うためのパッケージはいくつかありますが今回はDEseq2パッケージを使用します。 DEseq2パッケージの読み込み # ライブラリの読み込み
library (DESeq2)DEseq2パッケージはRaw Countsデータを使用します 。 正規化やlog変換をしたデータは渡してはいけません。 DESeqDataSetFromMatrixを使用してDESeq2オブジェクトを作成 # DESeq2オブジェクトを作成
dds <- DESeqDataSetFromMatrix (
countData = counts_mrna_matrix,
colData = metadata_sample_type,
design = ~ sample_type
) converting counts to integer mode
Warning in DESeqDataSet (se, design = design, ignoreRank): some variables in
design formula are characters, converting to factors class: DESeqDataSet
dim: 19938 100
metadata (1 ): version
assays (1 ): counts
rownames (19938 ): A1BG A1CF ... ZZEF1 ZZZ3
rowData names (0 ):
colnames (100 ): TCGA- D5-5540 TCGA- EI-6509 ... GTEX-139 YR-2126 - SM-5 KM11
GTEX-13 VXT-2426 - SM-5L U46
colData names (2 ): case_id sample_type 今回は事前フィルタリングを行いませんでした。 実はこの後の処理でDEseq2は自動的にフィルタリング を行ってくれます。 事前に緩い条件でフィルタリングしておくとデータが小さくなって計算しやすくなります。 今回は自動フィルタリングの挙動を見てもらうためにあえて事前フィルタリングを行いませんでした。 DESeq関数を使用してDEG解析を実行 # DEG解析の実行
dds <- DESeq (dds) estimating size factors
estimating dispersions
gene- wise dispersion estimates
mean- dispersion relationship
final dispersion estimates
fitting model and testing
-- replacing outliers and refitting for 1098 genes
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts (dds)
estimating dispersions
fitting model and testing class: DESeqDataSet
dim: 19938 100
metadata (1 ): version
assays (6 ): counts mu ... replaceCounts replaceCooks
rownames (19938 ): A1BG A1CF ... ZZEF1 ZZZ3
rowData names (23 ): baseMean baseVar ... maxCooks replace
colnames (100 ): TCGA- D5-5540 TCGA- EI-6509 ... GTEX-139 YR-2126 - SM-5 KM11
GTEX-13 VXT-2426 - SM-5L U46
colData names (4 ): case_id sample_type sizeFactor replaceable str(dds)の結果(長いので折り畳み) Formal class 'DESeqDataSet' [package "DESeq2" ] with 8 slots
..@ design : Class 'formula' language ~ sample_type
.. .. ..- attr (* , ".Environment" )=< environment: R_GlobalEnv>
..@ dispersionFunction: function (q)
.. ..- attr (* , "srcref" )= 'srcref' int [1: 8 ] 2212 11 2212 45 11 45 2774 2774
.. .. ..- attr (* , "srcfile" )= Classes 'srcfilealias' , 'srcfile' < environment: 0x57e774067cd0 >
.. ..- attr (* , "coefficients" )= Named num [1: 2 ] 0.43 13.84
.. .. ..- attr (* , "names" )= chr [1: 2 ] "asymptDisp" "extraPois"
.. ..- attr (* , "fitType" )= chr "parametric"
.. ..- attr (* , "varLogDispEsts" )= num 1.1
.. ..- attr (* , "dispPriorVar" )= num 1.08
..@ rowRanges : Formal class 'CompressedGRangesList' [package "GenomicRanges" ] with 5 slots
.. .. ..@ unlistData : Formal class 'GRanges' [package "GenomicRanges" ] with 7 slots
.. .. .. .. ..@ seqnames : Formal class 'Rle' [package "S4Vectors" ] with 4 slots
.. .. .. .. .. .. ..@ values : Factor w/ 0 levels:
.. .. .. .. .. .. ..@ lengths : int (0 )
.. .. .. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. .. .. ..@ metadata : list ()
.. .. .. .. ..@ ranges : Formal class 'IRanges' [package "IRanges" ] with 6 slots
.. .. .. .. .. .. ..@ start : int (0 )
.. .. .. .. .. .. ..@ width : int (0 )
.. .. .. .. .. .. ..@ NAMES : NULL
.. .. .. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. .. .. ..@ metadata : list ()
.. .. .. .. ..@ strand : Formal class 'Rle' [package "S4Vectors" ] with 4 slots
.. .. .. .. .. .. ..@ values : Factor w/ 3 levels "+" ,"-" ,"*" :
.. .. .. .. .. .. ..@ lengths : int (0 )
.. .. .. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. .. .. ..@ metadata : list ()
.. .. .. .. ..@ seqinfo : Formal class 'Seqinfo' [package "GenomeInfoDb" ] with 4 slots
.. .. .. .. .. .. ..@ seqnames : chr (0 )
.. .. .. .. .. .. ..@ seqlengths : int (0 )
.. .. .. .. .. .. ..@ is_circular: logi (0 )
.. .. .. .. .. .. ..@ genome : chr (0 )
.. .. .. .. ..@ elementMetadata: Formal class 'DFrame' [package "S4Vectors" ] with 6 slots
.. .. .. .. .. .. ..@ rownames : NULL
.. .. .. .. .. .. ..@ nrows : int 0
.. .. .. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. .. .. ..@ metadata : list ()
.. .. .. .. .. .. ..@ listData : Named list ()
.. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. ..@ metadata : list ()
.. .. ..@ elementMetadata: Formal class 'DFrame' [package "S4Vectors" ] with 6 slots
.. .. .. .. ..@ rownames : NULL
.. .. .. .. ..@ nrows : int 19938
.. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. ..@ elementMetadata: Formal class 'DFrame' [package "S4Vectors" ] with 6 slots
.. .. .. .. .. .. ..@ rownames : NULL
.. .. .. .. .. .. ..@ nrows : int 23
.. .. .. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. .. .. ..@ metadata : list ()
.. .. .. .. .. .. ..@ listData : List of 2
.. .. .. .. .. .. .. ..$ type : chr [1: 23 ] "intermediate" "intermediate" "intermediate" "intermediate" ...
.. .. .. .. .. .. .. ..$ description: chr [1: 23 ] "mean of normalized counts for all samples" "variance of normalized counts for all samples" "all counts for a gene are zero" "gene-wise estimates of dispersion" ...
.. .. .. .. ..@ metadata : list ()
.. .. .. .. ..@ listData : List of 23
.. .. .. .. .. ..$ baseMean : num [1: 19938 ] 57.63 945.03 22969.76 15.66 2.31 ...
.. .. .. .. .. ..$ baseVar : num [1: 19938 ] 4.86e+03 1.43e+06 3.43e+08 4.55e+02 8.60 ...
.. .. .. .. .. ..$ allZero : logi [1: 19938 ] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. .. .. .. ..$ dispGeneEst : num [1: 19938 ] 0.275 2.442 0.45 1.01 1.142 ...
.. .. .. .. .. ..$ dispGeneIter : num [1: 19938 ] 13 12 11 15 7 13 13 12 15 15 ...
.. .. .. .. .. ..$ dispFit : num [1: 19938 ] 0.67 0.445 0.431 1.313 6.419 ...
.. .. .. .. .. ..$ dispersion : num [1: 19938 ] 0.284 2.389 0.389 1.015 1.228 ...
.. .. .. .. .. ..$ dispIter : num [1: 19938 ] 11 11 12 12 12 9 8 12 10 11 ...
.. .. .. .. .. ..$ dispOutlier : logi [1: 19938 ] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. .. .. .. ..$ dispMAP : num [1: 19938 ] 0.284 2.389 0.389 1.015 1.228 ...
.. .. .. .. .. ..$ Intercept : num [1: 19938 ] 6.81 9.39 14.99 4.23 1.48 ...
.. .. .. .. .. ..$ sample_type_tumor_vs_normal : num [1: 19938 ] -5.136 0.863 -1.29 -0.564 -0.63 ...
.. .. .. .. .. ..$ SE_Intercept : num [1: 19938 ] 0.11 0.315 0.127 0.21 0.25 ...
.. .. .. .. .. ..$ SE_sample_type_tumor_vs_normal : num [1: 19938 ] 0.202 0.446 0.18 0.301 0.38 ...
.. .. .. .. .. ..$ WaldStatistic_Intercept : num [1: 19938 ] 61.89 29.77 117.84 20.15 5.92 ...
.. .. .. .. .. ..$ WaldStatistic_sample_type_tumor_vs_normal: num [1: 19938 ] -25.49 1.93 -7.17 -1.87 -1.66 ...
.. .. .. .. .. ..$ WaldPvalue_Intercept : num [1: 19938 ] 0.00 1.02e-194 0.00 2.86e-90 3.19e-09 ...
.. .. .. .. .. ..$ WaldPvalue_sample_type_tumor_vs_normal : num [1: 19938 ] 2.83e-143 5.31e-02 7.68e-13 6.13e-02 9.71e-02 ...
.. .. .. .. .. ..$ betaConv : logi [1: 19938 ] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. ..$ betaIter : num [1: 19938 ] 5 15 4 6 5 4 6 3 3 11 ...
.. .. .. .. .. ..$ deviance : num [1: 19938 ] 771 1493 2151 756 408 ...
.. .. .. .. .. ..$ maxCooks : logi [1: 19938 ] NA NA NA NA NA NA ...
.. .. .. .. .. ..$ replace : Named logi [1: 19938 ] FALSE FALSE FALSE TRUE FALSE FALSE ...
.. .. .. .. .. .. ..- attr (* , "names" )= chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. ..@ elementType : chr "GRanges"
.. .. ..@ metadata : list ()
.. .. ..@ partitioning : Formal class 'PartitioningByEnd' [package "IRanges" ] with 5 slots
.. .. .. .. ..@ end : int [1: 19938 ] 0 0 0 0 0 0 0 0 0 0 ...
.. .. .. .. ..@ NAMES : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. ..@ metadata : list ()
..@ colData : Formal class 'DFrame' [package "S4Vectors" ] with 6 slots
.. .. ..@ rownames : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. ..@ nrows : int 100
.. .. ..@ elementType : chr "ANY"
.. .. ..@ elementMetadata: Formal class 'DFrame' [package "S4Vectors" ] with 6 slots
.. .. .. .. ..@ rownames : NULL
.. .. .. .. ..@ nrows : int 4
.. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. ..@ metadata : list ()
.. .. .. .. ..@ listData : List of 2
.. .. .. .. .. ..$ type : chr [1: 4 ] "input" "input" "intermediate" "intermediate"
.. .. .. .. .. ..$ description: chr [1: 4 ] "" "" "a scaling factor for columns" "outliers can be replaced"
.. .. ..@ metadata : list ()
.. .. ..@ listData : List of 4
.. .. .. ..$ case_id : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. ..$ sample_type: Factor w/ 2 levels "normal" ,"tumor" : 2 2 2 2 2 2 2 2 2 2 ...
.. .. .. ..$ sizeFactor : Named num [1: 100 ] 0.661 1.019 1.051 1.487 1.367 ...
.. .. .. .. ..- attr (* , "names" )= chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. ..$ replaceable: logi [1: 100 ] TRUE TRUE TRUE TRUE TRUE TRUE ...
..@ assays : Formal class 'SimpleAssays' [package "SummarizedExperiment" ] with 1 slot
.. .. ..@ data: Formal class 'SimpleList' [package "S4Vectors" ] with 4 slots
.. .. .. .. ..@ listData : List of 6
.. .. .. .. .. ..$ counts : int [1: 19938 , 1 : 100 ] 1 786 2342 1 0 48 0 1898 1404 33 ...
.. .. .. .. .. .. ..- attr (* , "dimnames" )= List of 2
.. .. .. .. .. .. .. ..$ : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. .. .. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. .. .. ..$ mu : num [1: 19938 , 1 : 100 ] 2.11 806.18 8816.92 115.82 1.19 ...
.. .. .. .. .. .. ..- attr (* , "dimnames" )= List of 2
.. .. .. .. .. .. .. ..$ : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. .. .. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. .. .. ..$ H : num [1: 19938 , 1 : 100 ] 0.0181 0.02 0.02 0.0198 0.0189 ...
.. .. .. .. .. .. ..- attr (* , "dimnames" )= List of 2
.. .. .. .. .. .. .. ..$ : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. .. .. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. .. .. ..$ cooks : num [1: 19938 , 1 : 100 ] 2.26e-03 7.48e-06 1.08e-02 2.09e-01 7.36e-03 ...
.. .. .. .. .. .. ..- attr (* , "dimnames" )= List of 2
.. .. .. .. .. .. .. ..$ : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. .. .. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. .. .. ..$ replaceCounts: int [1: 19938 , 1 : 100 ] 1 786 2342 1 0 48 0 1898 1404 33 ...
.. .. .. .. .. .. ..- attr (* , "dimnames" )= List of 2
.. .. .. .. .. .. .. ..$ : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. .. .. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. .. .. ..$ replaceCooks : num [1: 19938 , 1 : 100 ] 2.26e-03 7.48e-06 1.08e-02 2.09e-01 7.36e-03 ...
.. .. .. .. .. .. ..- attr (* , "dimnames" )= List of 2
.. .. .. .. .. .. .. ..$ : chr [1: 19938 ] "A1BG" "A1CF" "A2M" "A2ML1" ...
.. .. .. .. .. .. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. .. .. ..@ elementType : chr "ANY"
.. .. .. .. ..@ elementMetadata: NULL
.. .. .. .. ..@ metadata : list ()
..@ NAMES : NULL
..@ elementMetadata : Formal class 'DFrame' [package "S4Vectors" ] with 6 slots
.. .. ..@ rownames : NULL
.. .. ..@ nrows : int 19938
.. .. ..@ elementType : chr "ANY"
.. .. ..@ elementMetadata: NULL
.. .. ..@ metadata : list ()
.. .. ..@ listData : Named list ()
..@ metadata : List of 1
.. ..$ version: Classes 'package_version' , 'numeric_version' hidden list of 1
.. .. ..$ : int [1: 3 ] 1 46 0
..$ betaPrior : logi FALSE
..$ modelMatrixType: chr "standard"
..$ betaPriorVar : num [1: 2 ] 1e+06 1e+06
..$ modelMatrix : num [1: 100 , 1 : 2 ] 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr (* , "dimnames" )= List of 2
.. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. ..$ : chr [1: 2 ] "Intercept" "sample_type_tumor_vs_normal"
.. ..- attr (* , "assign" )= int [1: 2 ] 0 1
.. ..- attr (* , "contrasts" )= List of 1
.. .. ..$ sample_type: chr "contr.treatment"
..$ test : chr "Wald"
..$ dispModelMatrix: num [1: 100 , 1 : 2 ] 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr (* , "dimnames" )= List of 2
.. .. ..$ : chr [1: 100 ] "TCGA-D5-5540" "TCGA-EI-6509" "TCGA-A6-6137" "TCGA-QG-A5Z2" ...
.. .. ..$ : chr [1: 2 ] "(Intercept)" "sample_typetumor"
.. ..- attr (* , "assign" )= int [1: 2 ] 0 1
.. ..- attr (* , "contrasts" )= List of 1
.. .. ..$ sample_type: chr "contr.treatment" DESeq関数の中では前回行った正規化やlog変換を自動的にやってくれています。 assaysとrowDataとcolDataがかなり増えたことが分かると思います。DESeq()関数はこれらを計算してくれました。 詳細についてはstr(dds)の結果をAIに解説してもらいましょう。 results関数を使用してDEG解析の結果を取得しサマリを確認 # 結果の取得
res <- results (
dds,
contrast = c ("sample_type" , "tumor" , "normal" ),
alpha = 0.01
)
# 結果の確認
summary (res)out of 19462 with nonzero total read count
adjusted p- value < 0.01
LFC > 0 (up) : 6746 , 35 %
LFC < 0 (down) : 6279, 32%
outliers [1] : 0 , 0 %
low counts [2] : 0, 0%
(mean count < 0 )
[1] see 'cooksCutoff' argument of ? results
[2] see 'independentFiltering' argument of ? results log2 fold change (MLE): sample_type tumor vs normal
Wald test p- value: sample type tumor vs normal
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
< numeric> < numeric> < numeric> < numeric> < numeric>
A1BG 57.62893 -5.135784 0.201515 -25.48582 2.83137e-143
A1CF 945.03499 0.862637 0.446072 1.93385 5.31316e-02
A2M 22969.76208 -1.289614 0.179944 -7.16675 7.67991e-13
A2ML1 15.66372 -0.563762 0.301272 -1.87127 6.13074e-02
A3GALT2 2.31026 -0.629796 0.379575 -1.65921 9.70729e-02
A4GALT 1179.80523 -1.738136 0.232010 -7.49164 6.80179e-14
padj
< numeric>
A1BG 2.96259e-141
A1CF 7.10686e-02
A2M 2.31623e-12
A2ML1 8.13336e-02
A3GALT2 1.25015e-01
A4GALT 2.16939e-13 res$padjのNA値とそうでない値の数を確認 # NA値の確認
table (is.na (res$ padj))NAになった遺伝子のbaseMeanを確認 # NAになった遺伝子のbaseMeanを確認
na_genes_basemean <- res[is.na (res$ padj), "baseMean" ]
summary (na_genes_basemean) Min. 1 st Qu. Median Mean 3 rd Qu. Max.
0 0 0 0 0 0 フィルタリングを行ったのでadjusted p-valueがNAになった遺伝子が出てきました。 今回は全サンプルでカウントが0の遺伝子 が統計的な処理に値しないため除去されています。 0ではないがカウントが低い遺伝子は今回除去されませんでした。 さらに厳しい条件でフィルタリングを行う場合はresults()関数のfilterパラメータを使用します。 results関数を使用してさらに平均遺伝子が10以上の遺伝子を抽出するフィルタリングを行う res_filter <- results (
dds,
contrast = c ("sample_type" , "tumor" , "normal" ),
alpha = 0.01 ,
filter = rowMeans (counts (dds, normalized = TRUE )) >= 10
)
# 結果の確認
summary (res_filter)out of 19462 with nonzero total read count
adjusted p- value < 0.01
LFC > 0 (up) : 6211 , 32 %
LFC < 0 (down) : 5954, 31%
outliers [1] : 0 , 0 %
low counts [2] : 3067, 16%
(mean count < 1 )
[1] see 'cooksCutoff' argument of ? results
[2] see 'independentFiltering' argument of ? results log2 fold change (MLE): sample_type tumor vs normal
Wald test p- value: sample type tumor vs normal
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
< numeric> < numeric> < numeric> < numeric> < numeric>
A1BG 57.62893 -5.135784 0.201515 -25.48582 2.83137e-143
A1CF 945.03499 0.862637 0.446072 1.93385 5.31316e-02
A2M 22969.76208 -1.289614 0.179944 -7.16675 7.67991e-13
A2ML1 15.66372 -0.563762 0.301272 -1.87127 6.13074e-02
A3GALT2 2.31026 -0.629796 0.379575 -1.65921 9.70729e-02
A4GALT 1179.80523 -1.738136 0.232010 -7.49164 6.80179e-14
padj
< numeric>
A1BG 2.49571e-141
A1CF 6.54514e-02
A2M 2.01330e-12
A2ML1 7.50213e-02
A3GALT2 NA
A4GALT 1.88180e-13 res_filter$padjのNA値とそうでない値の数を確認 # NA値の確認
table (is.na (res_filter$ padj))NAになった遺伝子のbaseMeanを確認 # NA値の遺伝子のbaseMeanを確認
na_genes_basemean_filter <- res_filter[is.na (res_filter$ padj), "baseMean" ]
summary (na_genes_basemean_filter) Min. 1 st Qu. Median Mean 3 rd Qu. Max.
0.0000 0.1574 0.7521 1.8663 2.6059 9.9877 このように正規化されたカウントが10以上の遺伝子のみが残りました。 カウントが低い遺伝子は偽陽性率が上がりやすくなるため除去すると統計的な信頼性が上がります。 しかし、カウントが低い遺伝子に注目したい研究の場合は意図的に緩くすることも必要になってきます。 今回は厳しめにフィルタリングしたデータを使って後続の解析をしましょう。 res_filterをtibbleに変換 # tidyverseで操作しやすいようにresをtibbleに変換
res_tibble <- res_filter %>%
as.data.frame () %>%
rownames_to_column ("gene_name" ) %>%
as_tibble ()
head (res_tibble)# A tibble: 6 × 7
gene_name baseMean log2FoldChange lfcSE stat pvalue padj
< chr> < dbl> < dbl> < dbl> < dbl> < dbl> < dbl>
1 A1BG 57.6 -5.14 0.202 -25.5 2.83e-143 2.50e-141
2 A1CF 945 . 0.863 0.446 1.93 5.31 e- 2 6.55 e- 2
3 A2M 22970 . -1.29 0.180 -7.17 7.68 e- 13 2.01 e- 12
4 A2ML1 15.7 -0.564 0.301 -1.87 6.13 e- 2 7.50 e- 2
5 A3GALT2 2.31 -0.630 0.380 -1.66 9.71 e- 2 NA
6 A4GALT 1180 . -1.74 0.232 -7.49 6.80 e- 14 1.88 e- 13 細かい結果を知りたい場合は結果のフィルタリングを行ってから表示します 。 adjusted p-valueが0.01未満の遺伝子を抽出 # adjusted p-valueが0.01未満の遺伝子を抽出
padj_threshold <- 0.01
res_filtered <- res_tibble %>%
filter (padj < padj_threshold)
print (head (res_filtered))# A tibble: 6 × 7
gene_name baseMean log2FoldChange lfcSE stat pvalue padj
< chr> < dbl> < dbl> < dbl> < dbl> < dbl> < dbl>
1 A1BG 57.6 -5.14 0.202 -25.5 2.83e-143 2.50e-141
2 A2M 22970 . -1.29 0.180 -7.17 7.68 e- 13 2.01 e- 12
3 A4GALT 1180 . -1.74 0.232 -7.49 6.80 e- 14 1.88 e- 13
4 AAAS 2687 . -0.678 0.0974 -6.96 3.33 e- 12 8.43 e- 12
5 AACS 1837 . 0.884 0.143 6.20 5.63 e- 10 1.26 e- 9
6 AADAC 29.3 1.62 0.464 3.49 4.86 e- 4 7.22 e- 4 結果を表示 print (paste0 ("adjusted p-valueが" , padj_threshold, "の時に有意なDEGの数: " , nrow (res_filtered)))
print (paste0 ("有意な遺伝子数の割合は" , nrow (res_filtered) / nrow (res_tibble) * 100 , "%" ))[1] "adjusted p-valueが0.01の時に有意なDEGの数: 12165" [1] "有意な遺伝子数の割合は61.0141438459224%" adjusted p-valueが0.01未満でlogFoldChange > 2 の遺伝子(up-regulated)を抽出 # adjusted p-valueが0.01未満でlogFoldChange > 2 の遺伝子(up-regulated)を抽出
res_filtered_up <- res_tibble %>%
filter (log2FoldChange > 2 & padj < padj_threshold) %>%
# logFCが大きい方から順番に並べる
arrange (desc (log2FoldChange))
print (head (res_filtered_up))# A tibble: 6 × 7
gene_name baseMean log2FoldChange lfcSE stat pvalue padj
< chr> < dbl> < dbl> < dbl> < dbl> < dbl> < dbl>
1 PRRC2B 3425 . 15.6 0.324 48.1 0 0
2 BX255925.3 473 . 12.7 0.335 38.0 0 0
3 F8A1 155 . 11.1 0.327 34.0 8.42e-254 1.82e-251
4 CST1 791 . 10.6 0.582 18.2 1.11 e- 73 2.88 e- 72
5 RTEL1- TNFRSF6B 85.5 10.3 0.373 27.5 1.90e-166 2.17e-164
6 NOTUM 3051 . 10.2 0.476 21.4 2.72e-101 1.31 e- 99 print (paste0 ("up-regulatedの遺伝子数: " , nrow (res_filtered_up)))
print (paste0 ("up-regulatedの遺伝子数の割合は" , nrow (res_filtered_up) / nrow (res_tibble) * 100 , "%" ))[1] "up-regulatedの遺伝子数: 1177" [1] "up-regulatedの遺伝子数の割合は5.90330023071522%" adjusted p-valueが0.01未満でlogFoldChange < -2 の遺伝子(down-regulated)を抽出 # adjusted p-valueが0.01未満でlogFoldChange < -2 の遺伝子(down-regulated)を抽出
res_filtered_down <- res_tibble %>%
filter (log2FoldChange < -2 & padj < padj_threshold) %>%
# logFCが小さい方から順番に並べる
arrange (log2FoldChange)
print (head (res_filtered_down))# A tibble: 6 × 7
gene_name baseMean log2FoldChange lfcSE stat pvalue padj
< chr> < dbl> < dbl> < dbl> < dbl> < dbl> < dbl>
1 EEF1G 21738 . -17.2 0.293 -58.8 0 0
2 TOMM6 1295 . -13.4 0.294 -45.7 0 0
3 NICN1 944 . -13.0 0.303 -42.7 0 0
4 WDR83OS 863 . -12.4 0.294 -42.3 0 0
5 UPK3BL1 467 . -11.9 0.319 -37.4 7.25e-307 2.05e-304
6 TNFRSF6B 437 . -11.8 0.366 -32.3 1.53e-229 2.67e-227 print (paste0 ("down-regulatedの遺伝子数: " , nrow (res_filtered_down)))
print (paste0 ("down-regulatedの遺伝子数の割合は" , nrow (res_filtered_down) / nrow (res_tibble) * 100 , "%" ))[1] "down-regulatedの遺伝子数: 2025" [1] "down-regulatedの遺伝子数の割合は10.1564851038218%" 3. Volcano plotを作ってDEGの分布を視覚的に捉える。
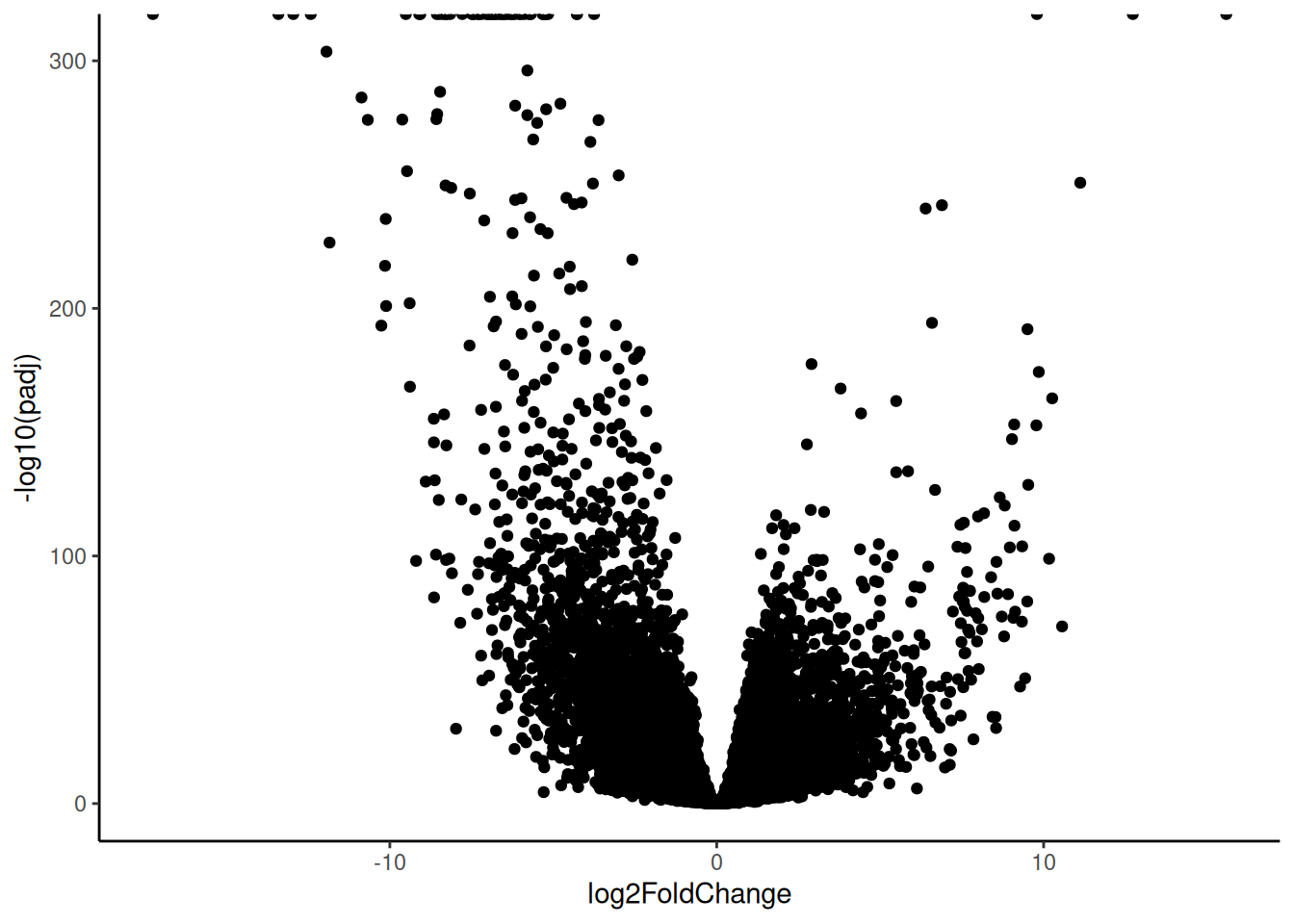
# DEGの結果をグラフで可視化してみます。 log2FC(Fold Change)の絶対値の大きさは遺伝子の発現量の変化の大きさを表しています。 adjusted p-valueが小さい遺伝子ほど統計学的に有意になりやすい遺伝子です。 一般的にlog2FCの絶対値が大きくて有意な遺伝子が特に重要な遺伝子と考えられます。 そこでlog2FCとadjusted p-valueの値を使って書いたグラフをVolcano plotといいます。 ggplotでvolcano plotを作成 # グラフの作成
ggplot (res_tibble, aes (x = log2FoldChange, y = - log10 (padj))) +
geom_point () +
theme_classic () Warning: Removed 3543 rows containing missing values or values outside the scale range
`geom_point ()`). warningはどうすればよい?
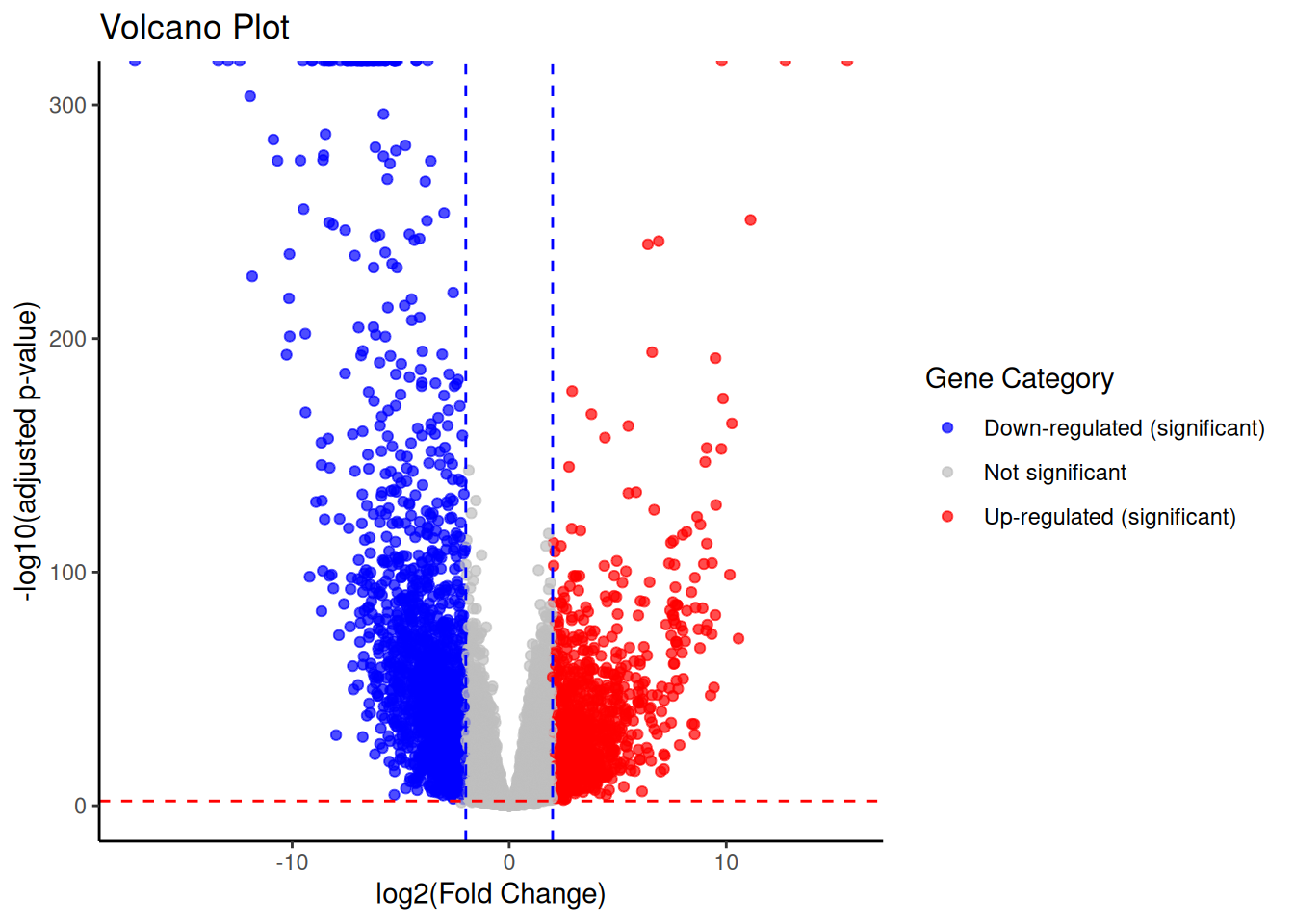
# コードに致命的な間違いがある場合はerrorが起きてコードは実行されません。 一方でコード自体は動くが、想定通りの働きをしていない可能性がある場合にwarning が起きます。 warningの場合、コードは進みますし実際のところ問題ない場合も多いです。 しかし、結果の解釈に大きな影響を与えるwarningも存在し、特にwarningが発生した図を他の人に見せる時 には注意が必要です。 まずはwarningの意味をAIに聞いてしっかり読んでみましょう。 このようにadjusted p-valueに負号をつけることによって、小さいほど上に表示されるようにします。 ということは、例えばadjusted p-value < 0.01の遺伝子を有意と判定することにした場合、上図の-log10(0.01)の値よりも上の遺伝子が有意ということになります。 そのような平行線を書き加えて、有意な遺伝子の色を変えてみましょう。 またlog2FCの絶対値が大きい遺伝子も知りたいので、今回はlog2FCの絶対値が2以上の遺伝子も垂直線で示して色を変えてみます。 Up-regulatedやDown-regulatedやNotsignificantのようなカテゴリ変数を作成して色分けの準備 # カテゴリ変数を作成して色分けの準備
res_tibble_plot <- res_tibble %>%
mutate (
significance = case_when (
padj < 0.01 & log2FoldChange > 2 ~ "Up-regulated (significant)" ,
padj < 0.01 & log2FoldChange < -2 ~ "Down-regulated (significant)" ,
TRUE ~ "Not significant"
)
)
head (res_tibble_plot)# A tibble: 6 × 8
gene_name baseMean log2FoldChange lfcSE stat pvalue padj
< chr> < dbl> < dbl> < dbl> < dbl> < dbl> < dbl>
1 A1BG 57.6 -5.14 0.202 -25.5 2.83e-143 2.50e-141
2 A1CF 945 . 0.863 0.446 1.93 5.31 e- 2 6.55 e- 2
3 A2M 22970 . -1.29 0.180 -7.17 7.68 e- 13 2.01 e- 12
4 A2ML1 15.7 -0.564 0.301 -1.87 6.13 e- 2 7.50 e- 2
5 A3GALT2 2.31 -0.630 0.380 -1.66 9.71 e- 2 NA
6 A4GALT 1180 . -1.74 0.232 -7.49 6.80 e- 14 1.88 e- 13
# ℹ 1 more variable: significance <chr> table (res_tibble_plot$ significance)Down- regulated (significant) Not significant
2025 16736
Up- regulated (significant)
1177 ggplotでvolcano plotを作成(点の色分けとthresholdの線を追加) # グラフの作成
ggplot (res_tibble_plot, aes (x = log2FoldChange, y = - log10 (padj), color = significance)) +
geom_point (alpha = 0.7 ) +
geom_hline (yintercept = - log10 (0.01 ), color = "red" , linetype = "dashed" ) +
geom_vline (xintercept = c (-2 , 2 ), color = "blue" , linetype = "dashed" ) +
scale_color_manual (values = c (
"Up-regulated (significant)" = "red" ,
"Down-regulated (significant)" = "blue" ,
"Not significant" = "gray"
)) +
labs (
x = "log2(Fold Change)" ,
y = "-log10(adjusted p-value)" ,
color = "Gene Category" ,
title = "Volcano Plot"
) +
theme_classic () +
theme (legend.position = "right" ) Warning: Removed 3543 rows containing missing values or values outside the scale range
`geom_point ()`). さらにlogFCの絶対値が大きい上位10遺伝子のラベルを追加してみましょう。 # ライブラリの読み込み
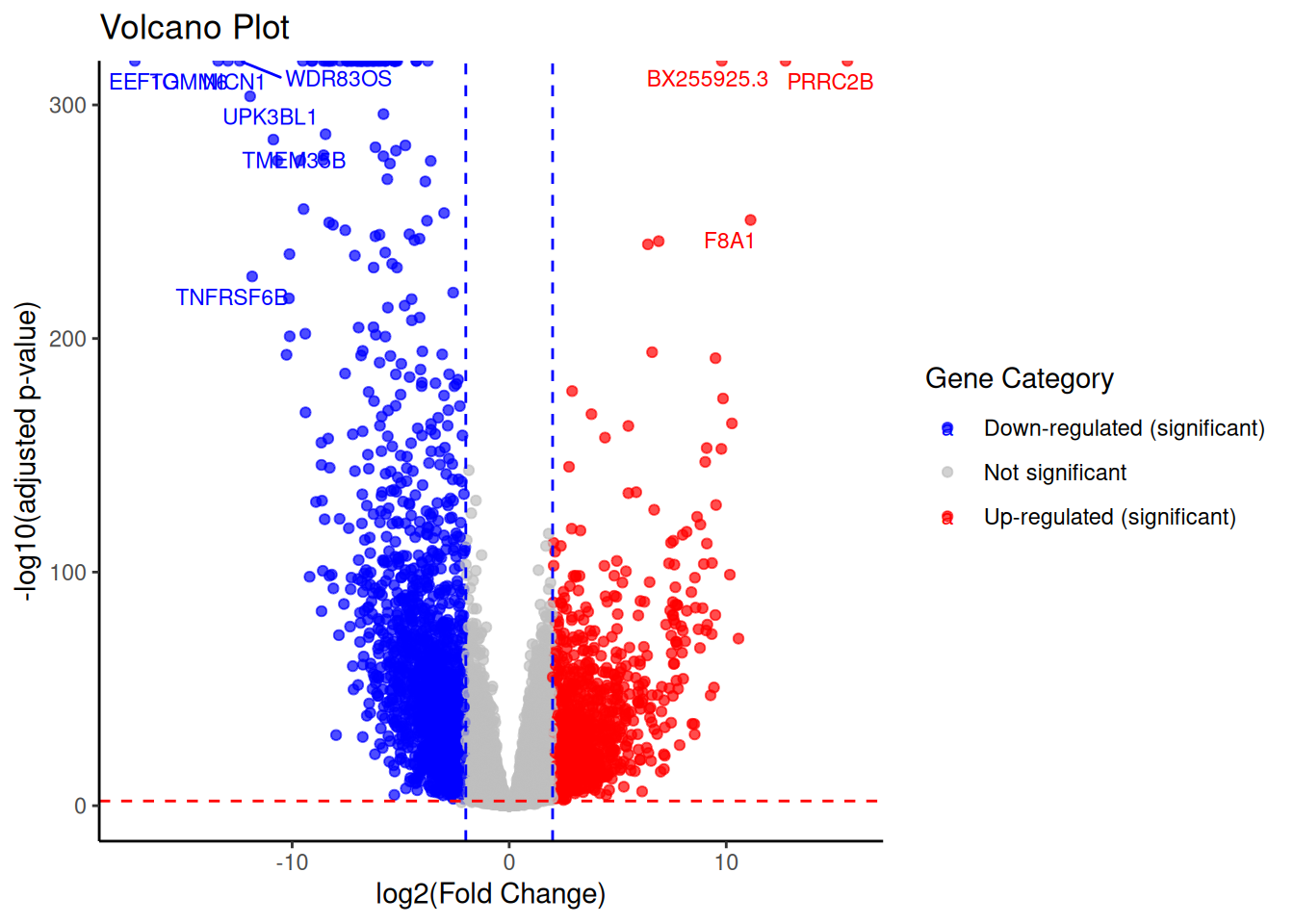
library (ggrepel)上位10遺伝子の情報を抽出 # 上位10遺伝子のラベルを追加
res_tibble_plot_label <- res_tibble_plot %>%
arrange (desc (abs (log2FoldChange))) %>%
head (10 )
print (res_tibble_plot_label)# A tibble: 10 × 8
gene_name baseMean log2FoldChange lfcSE stat pvalue padj
< chr> < dbl> < dbl> < dbl> < dbl> < dbl> < dbl>
1 EEF1G 21738 . -17.2 0.293 -58.8 0 0
2 PRRC2B 3425 . 15.6 0.324 48.1 0 0
3 TOMM6 1295 . -13.4 0.294 -45.7 0 0
4 NICN1 944 . -13.0 0.303 -42.7 0 0
5 BX255925.3 473 . 12.7 0.335 38.0 0 0
6 WDR83OS 863 . -12.4 0.294 -42.3 0 0
7 UPK3BL1 467 . -11.9 0.319 -37.4 7.25e-307 2.05e-304
8 TNFRSF6B 437 . -11.8 0.366 -32.3 1.53e-229 2.67e-227
9 F8A1 155 . 11.1 0.327 34.0 8.42e-254 1.82e-251
10 TMEM35B 389 . -10.9 0.299 -36.3 2.54e-288 6.84e-286
# ℹ 1 more variable: significance <chr> ggplotでvolcano plotを作成(上位10遺伝子のラベルを追加。ggrepelライブラリを使用) ggplot (res_tibble_plot, aes (x = log2FoldChange, y = - log10 (padj), color = significance)) +
geom_point (alpha = 0.7 ) +
geom_text_repel (data = res_tibble_plot_label, aes (label = gene_name), size = 3 ) +
geom_hline (yintercept = - log10 (0.01 ), color = "red" , linetype = "dashed" ) +
geom_vline (xintercept = c (-2 , 2 ), color = "blue" , linetype = "dashed" ) +
scale_color_manual (values = c (
"Up-regulated (significant)" = "red" ,
"Down-regulated (significant)" = "blue" ,
"Not significant" = "gray"
)) +
labs (
x = "log2(Fold Change)" ,
y = "-log10(adjusted p-value)" ,
color = "Gene Category" ,
title = "Volcano Plot"
) +
theme_classic () +
theme (legend.position = "right" ) Warning: Removed 3543 rows containing missing values or values outside the scale range
`geom_point ()`). 大腸がんと正常大腸の間で変動のある遺伝子を網羅的に解析し可視化することができました。 4. パスウェイ解析
# 大腸がんと正常大腸の間で変動のある遺伝子を網羅的に解析し可視化することができました。 これらの遺伝子がどのようなパスウェイを介して関わっているかを解析することができます。 このような解析を行うためのパッケージはいくつかありますが今回はclusterProfilerパッケージを使用します。 また、今回はGO:BP というデータセットに登録されているパスウェイをつかって解析します。 GO:BPとは
# GO(Gene Ontology) :遺伝子の機能を体系的に分類するデータベースBP(Biological Process) :生物学的プロセス(細胞分裂、炎症反応など)その他のデータベース :KEGG、Reactome、MSigDBなど他にも目的に合わせて適切なデータベースがあります。# パスウェイ解析に必要なライブラリを読み込み
library (clusterProfiler)
library (org.Hs.eg.db)
library (enrichplot)ORA(Over-Representation Analysis)
# まずは有意に発現変動した遺伝子のみ を使用してパスウェイ解析を行います。 このような手法をORA(Over-Representation Analysis)といいます。 それぞれのパスウェイに含まれるgene setに、今回有意になった遺伝子がどのくらい含まれているかを検定します。 まずはパスウェイ解析に使用する遺伝子リストを決定します。 res_tibbleにはフィルタリングによってadjusted p-valueがNAになっている遺伝子が含まれているので除去しましょう。 padjがNAになっている遺伝子を除去して残った遺伝子の数を確認 # フィルタリングされた遺伝子を除去
universe_genes <- res_tibble %>%
filter (! is.na (padj)) %>%
pull (gene_name)
length (universe_genes)遺伝子名をEntrez IDに変換 # 遺伝子名をEntrez IDに変換
universe_genes_entrez <- clusterProfiler:: bitr (
geneID = universe_genes,
fromType = "SYMBOL" ,
toType = "ENTREZID" ,
OrgDb = org.Hs.eg.db
) 'select()' returned 1 : many mapping between keys and columns
Warning in clusterProfiler:: bitr (geneID = universe_genes, fromType = "SYMBOL" ,
: 1.64 % of input gene IDs are fail to map...head (universe_genes_entrez)
length (universe_genes_entrez$ ENTREZID) SYMBOL ENTREZID
1 A1BG 1
2 A1CF 29974
3 A2M 2
4 A2ML1 144568
5 A4GALT 53947
6 AAAS 8086 Entrez IDに変換できなかった遺伝子の数を確認 # Entrez IDに変換できなかった遺伝子の数
print (paste0 ("Entrez IDに変換できなかった遺伝子の数: " , length (universe_genes) - length (universe_genes_entrez$ ENTREZID), "遺伝子" ))[1] "Entrez IDに変換できなかった遺伝子の数: 268遺伝子" 一部の遺伝子はEntrez IDが対応していません。 対応していない遺伝子はそもそもパスウェイ内での機能が不明な遺伝子です。 今回の解析に含めないのでこのまま進んで問題ありません。 logFCとadjusted p-valueの値を結合して有意な遺伝子を抽出できるようにします。 universe_genes_entrezにres_tibbleを結合 # universe_genes_entrezにres_tibbleを結合
universe_genes_entrez_res <- universe_genes_entrez %>%
left_join (res_tibble, by = c ("SYMBOL" = "gene_name" ))
head (universe_genes_entrez_res) SYMBOL ENTREZID baseMean log2FoldChange lfcSE stat
1 A1BG 1 57.62893 -5.1357838 0.20151536 -25.485818
2 A1CF 29974 945.03499 0.8626370 0.44607241 1.933850
3 A2M 2 22969.76208 -1.2896141 0.17994406 -7.166750
4 A2ML1 144568 15.66372 -0.5637625 0.30127233 -1.871272
5 A4GALT 53947 1179.80523 -1.7381358 0.23201002 -7.491641
6 AAAS 8086 2686.95690 -0.6779640 0.09736328 -6.963242
pvalue padj
1 2.831369e-143 2.495715e-141
2 5.313162e-02 6.545142e-02
3 7.679907e-13 2.013305e-12
4 6.130740e-02 7.502126e-02
5 6.801787e-14 1.881797e-13
6 3.325306e-12 8.425034e-12 up-regulatedとdown-regulatedそれぞれで解析を行うためにフィルタリングを行います。 up-regulatedとdown-regulatedそれぞれで有意な遺伝子を抽出 # 有意な遺伝子を抽出(upregulated/downregulated別々に)
deg_genes_up <- universe_genes_entrez_res %>%
filter (padj < 0.01 & log2FoldChange > 2 )
deg_genes_down <- universe_genes_entrez_res %>%
filter (padj < 0.01 & log2FoldChange < -2 ) print (paste0 ("Upregulated遺伝子数: " , nrow (deg_genes_up), "遺伝子" ))
print (paste0 ("Downregulated遺伝子数: " , nrow (deg_genes_down), "遺伝子" ))[1] "Upregulated遺伝子数: 1134遺伝子" [1] "Downregulated遺伝子数: 2000遺伝子" 一般に有意な遺伝子が1000以上あれば十分に検定できるとされています。 ここまで用意できたらORAをGO:BPデータセットを使って実行します。 ここで重要なのが背景遺伝子セット の設定です。 これは通常、DEG解析でフィルタリングされた遺伝子を使用します。 つまり今回は正規化されたカウントが10以上の遺伝子を背景遺伝子セットとして使用します。 universe_genes_entrez_resはまさにこれらの遺伝子のリストなのでこれを利用します。 ORAの実行(up-regulatedとdown-regulatedそれぞれで実行)。適切な背景遺伝子を設定する。 # ORAの実行(up-regulated)
ora_result_up <- enrichGO (
gene = deg_genes_up$ ENTREZID,
universe = universe_genes_entrez_res$ ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "BP" ,
pAdjustMethod = "BH" ,
pvalueCutoff = 0.05 ,
qvalueCutoff = 0.05 ,
readable = TRUE
)
# ORAの実行(down-regulated)
ora_result_down <- enrichGO (
gene = deg_genes_down$ ENTREZID,
universe = universe_genes_entrez_res$ ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "BP" ,
pAdjustMethod = "BH" ,
pvalueCutoff = 0.05 ,
qvalueCutoff = 0.05 ,
readable = TRUE
) 結果の確認(p.adjust < 0.05のパスウェイ数をup-regulatedとdown-regulatedそれぞれで確認) # 結果の確認
print (paste0 ("Upregulated有意パスウェイ数: " , sum (ora_result_up@ result$ p.adjust < 0.05 ), "パスウェイ" ))
print (paste0 ("Downregulated有意パスウェイ数: " , sum (ora_result_down@ result$ p.adjust < 0.05 ), "パスウェイ" ))[1] "Upregulated有意パスウェイ数: 333パスウェイ" [1] "Downregulated有意パスウェイ数: 350パスウェイ" 有意なパスウェイを得ることができました。 背景遺伝子の設定がどのように行われているか確認してみましょう。 # 背景遺伝子数の確認(up-regulated)
bg_total_up <- as.numeric (strsplit (ora_result_up$ BgRatio[1], "/" )[[1]][2])
print (paste0 ("実際に使用された背景遺伝子数: " , bg_total_up))
print (paste0 ("入力した背景遺伝子数: " , length (universe_genes_entrez_res$ ENTREZID)))[1] "実際に使用された背景遺伝子数: 14765" # 背景遺伝子数の確認(down-regulated)
bg_total_down <- as.numeric (strsplit (ora_result_down$ BgRatio[1], "/" )[[1]][2])
print (paste0 ("実際に使用された背景遺伝子数: " , bg_total_down))[1] "実際に使用された背景遺伝子数: 14765" 背景遺伝子数は入力した遺伝子数よりも少ないことがわかります。 これはOrgDbデータベースに登録されていない遺伝子が自動的に除外されたためです。 データベースにないものはそもそも機能が不明でパスウェイに登録されていないということなので除外した方が良いです。 もしも背景遺伝子セットを設定しなかったら?
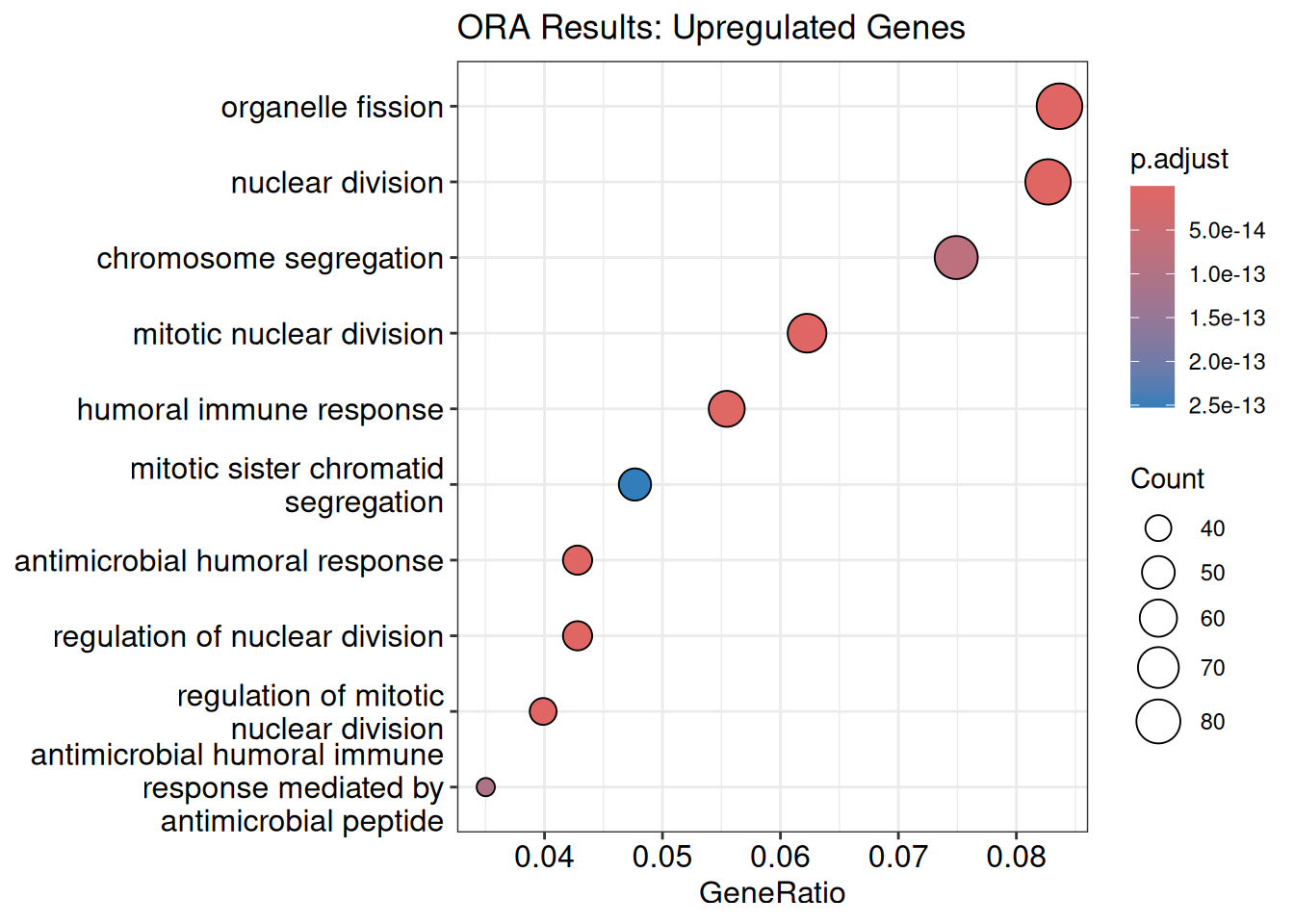
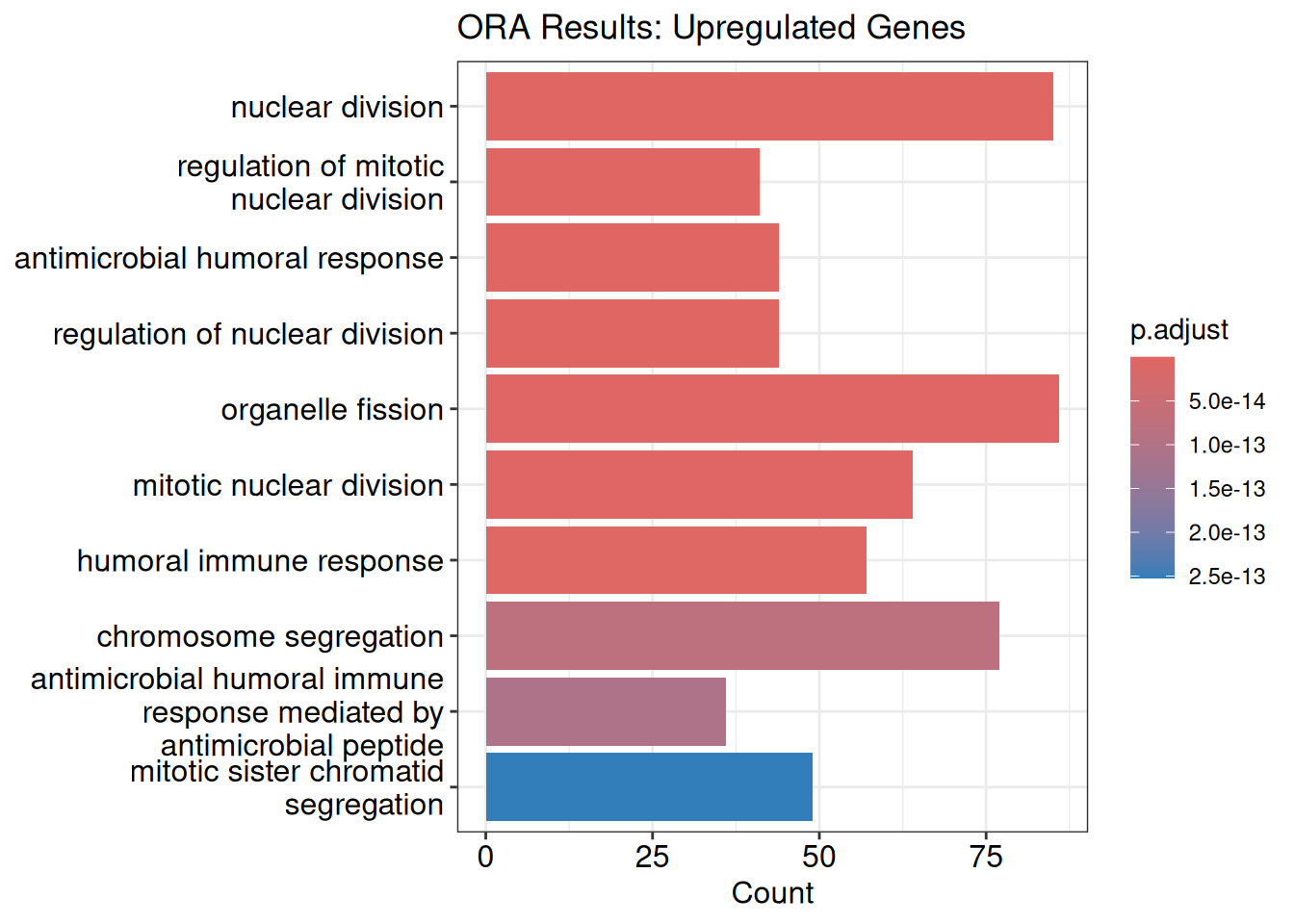
# universe引数になにも設定しなかった場合、背景遺伝子数は18,986遺伝子と増加します。 これは今回低カウントだったために除外された遺伝子も含んでいるためです。 背景遺伝子が多くなるとadjusted p-valueは小さく(有意になりやすく)なります。 しかし、実際は19000遺伝子近く解析しているわけではないので、偽陽性が増える 可能性が高いです。 次に結果を可視化します。 有意なパスウェイを10個表示してみます。 dotplotで可視化(up-regulated) # ORA結果の可視化
dotplot (ora_result_up, showCategory = 10 ) +
labs (title = "ORA Results: Upregulated Genes" ) barplotで可視化(up-regulated) barplot (ora_result_up, showCategory = 10 ) +
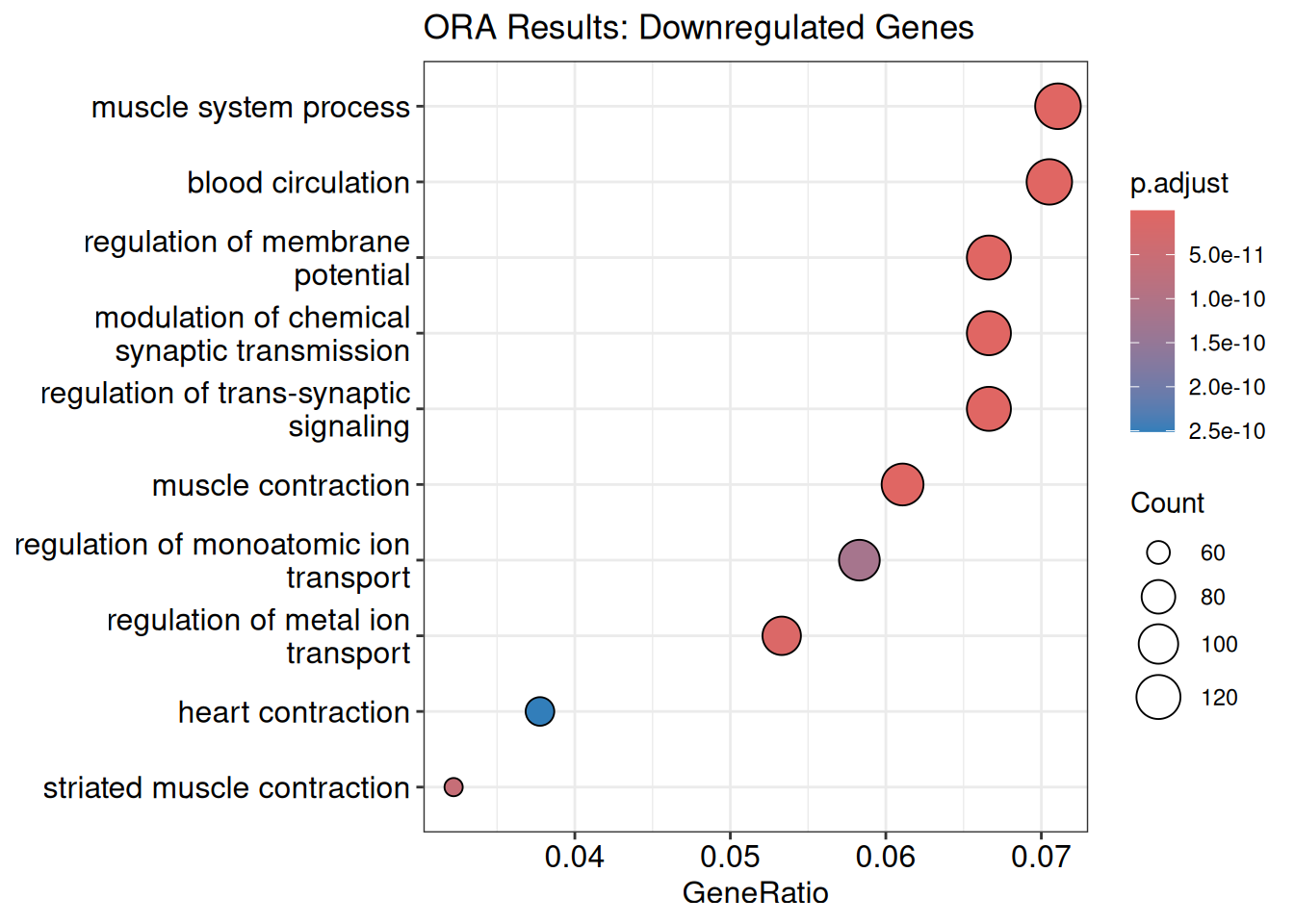
labs (title = "ORA Results: Upregulated Genes" ) dotplotで可視化(down-regulated) dotplot (ora_result_down, showCategory = 10 ) +
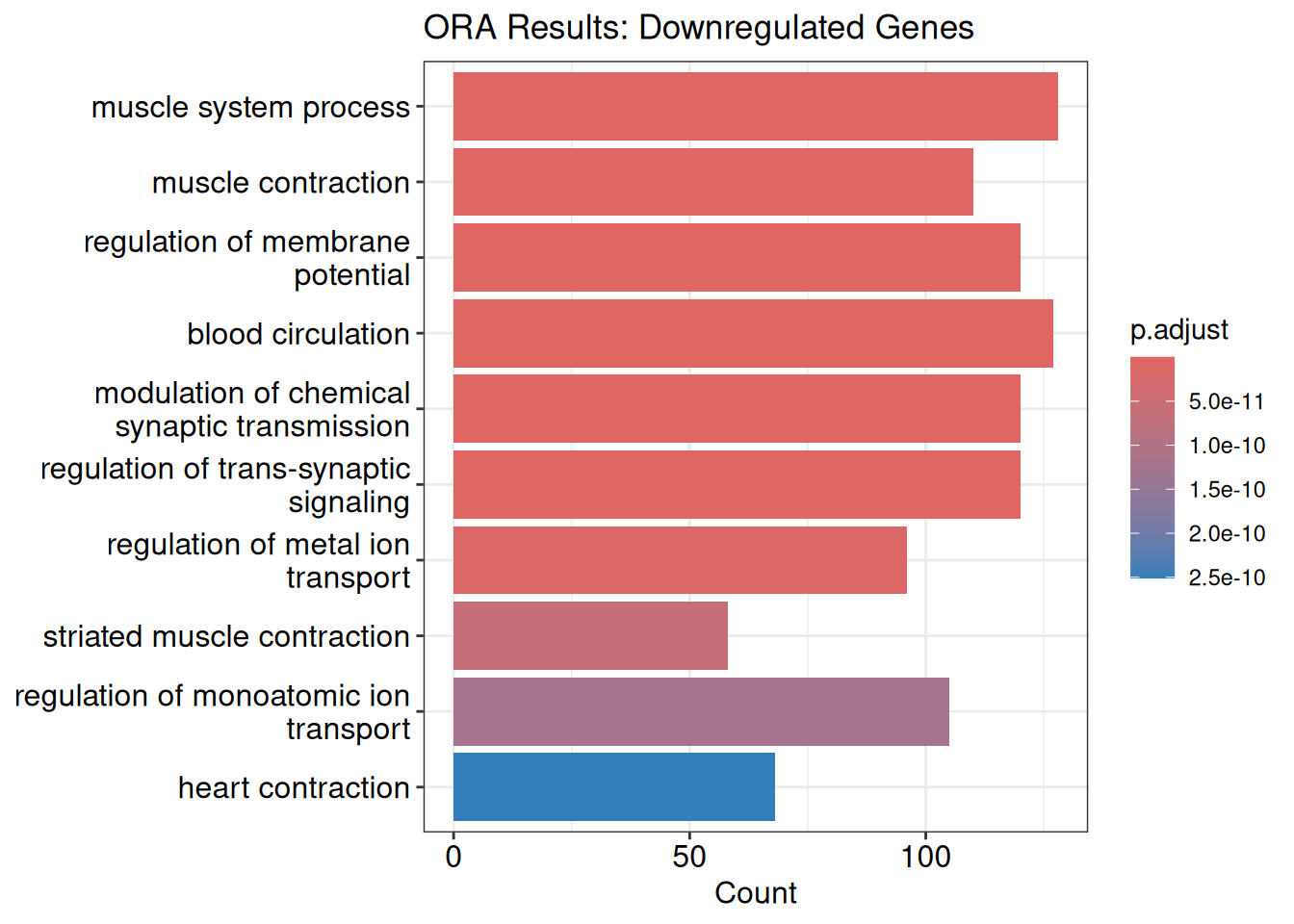
labs (title = "ORA Results: Downregulated Genes" ) barplotで可視化(down-regulated) barplot (ora_result_down, showCategory = 10 ) +
labs (title = "ORA Results: Downregulated Genes" ) このように自分で決めたthresholdを満たすパスウェイを一覧で表示することができました。 図の中の値が示す意味についてAIに聞いてみましょう。 GSEA(Gene Set Enrichment Analysis)
# 先ほどは有意な遺伝子のみを使用する方法でしたが、全遺伝子の情報を利用したパスウェイ解析もあります。 全遺伝子を何らかの統計量でランク付けして、その上位の遺伝子がどのパスウェイに多く含まれているかを検定します。 このような手法を**GSEA(Gene Set Enrichment Analysis)**といいます。 今回はlogFCの値を使ってランク付けを行います。 logFCがNAの遺伝子は除外してランク付けを行います。 なぜGSEAでは全遺伝子を使用するのか?
# 全遺伝子を使用することで、緩やかな変動を示す遺伝子セットも検出できるようになります。 例えば、logFCの値が0.1程度ではあるが、同じパスウェイに関連する遺伝子が何十個も同時に動いていたとします。このような遺伝子セットは、通常のORAでは検出できないことがあります。 GSEAであればこのような緩やかな変化が多くの遺伝子にわたって起きている ことを検出することができます。 ランク付けの方法
# logFC以外にもadjusted p-valueを使ったりその二つを組み合わせる手法などがあります。 そもそもp-valueの計算方法を変えたり、各群の平均や分散を用いて計算した統計量を使う方法などもあります。 それぞれの手法については後日別の解説を作成します。 全遺伝子のランク付けリストを作成(logFCを使用) # 全遺伝子のランク付けリストを作成
gene_list <- res_tibble %>%
filter (! is.na (log2FoldChange) & ! is.na (padj)) %>%
arrange (desc (log2FoldChange)) %>%
pull (log2FoldChange, name = gene_name) # 使用遺伝子数を確認
print ("使用遺伝子数:" , length (gene_list), "遺伝子" )遺伝子名をEntrez IDに変換 # 遺伝子名をEntrez IDに変換
gene_list_entrez <- clusterProfiler:: bitr (
geneID = names (gene_list),
fromType = "SYMBOL" ,
toType = "ENTREZID" ,
OrgDb = org.Hs.eg.db
) 'select()' returned 1 : many mapping between keys and columns
Warning in clusterProfiler:: bitr (geneID = names (gene_list), fromType =
"SYMBOL" , : 1.64 % of input gene IDs are fail to map...Entrez IDに変換できた遺伝子でランク付けリストを再作成 # マッチした遺伝子でランク付けリストを再作成
gene_list_filtered <- gene_list[gene_list_entrez$ SYMBOL]
names (gene_list_filtered) <- gene_list_entrez$ ENTREZID
gene_list_filtered <- sort (gene_list_filtered, decreasing = TRUE ) # Entrez IDに変換できた遺伝子数を確認
print (paste0 ("Entrez IDに変換できた遺伝子数: " , length (gene_list_filtered), "遺伝子" ))Entrez IDに変換できた遺伝子数: 16127 遺伝子 GSEA GO:BPを実行 # GSEAの実行
gse <- gseGO (
geneList = gene_list_filtered,
ont = "BP" ,
OrgDb = org.Hs.eg.db,
pvalueCutoff = 0.05 ,
pAdjustMethod = "BH" ,
verbose = FALSE
)
# 遺伝子名をEntrez IDからgene_nameに変換
gse <- setReadable (gse, OrgDb = org.Hs.eg.db, keyType = "ENTREZID" ) Warning in preparePathwaysAndStats (pathways, stats, minSize, maxSize, gseaParam, : There are ties in the preranked stats (0.01 % of the list).
The order of those tied genes will be arbitrary, which may produce unexpected results.
Warning in fgseaMultilevel (pathways = pathways, stats = stats, minSize =
minSize, : For some of the pathways the P- values were likely overestimated. For
such pathways log2err is set to NA.
Warning in fgseaMultilevel (pathways = pathways, stats = stats, minSize =
minSize, : For some pathways, in reality P- values are less than 1e-10 . You can
set the `eps` argument to zero for better estimation. ID Description setSize
GO: 0034502 GO: 0034502 protein localization to chromosome 119
GO: 0006261 GO: 0006261 DNA- templated DNA replication 160
GO: 0006260 GO: 0006260 DNA replication 268
GO: 0006936 GO: 0006936 muscle contraction 317
GO: 0001906 GO: 0001906 cell killing 207
GO: 0003012 GO: 0003012 muscle system process 402
enrichmentScore NES pvalue p.adjust qvalue rank
GO: 0034502 0.5769324 2.367958 1e-10 3.204211e-08 2.6e-08 2455
GO: 0006261 0.5410131 2.347688 1e-10 3.204211e-08 2.6e-08 2358
GO: 0006260 0.4761119 2.175801 1e-10 3.204211e-08 2.6e-08 2785
GO: 0006936 -0.5613291 -2.152419 1e-10 3.204211e-08 2.6e-08 2883
GO: 0001906 0.4815966 2.146887 1e-10 3.204211e-08 2.6e-08 2931
GO: 0003012 -0.5462766 -2.128093 1e-10 3.204211e-08 2.6e-08 3033
leading_edge
GO: 0034502 tags= 37 %, list=15% , signal= 32 %
GO:0006261 tags=41% , list= 15 %, signal=35%
GO: 0006260 tags= 36 %, list=17% , signal= 30 %
GO:0006936 tags=44% , list= 18 %, signal=37%
GO: 0001906 tags= 41 %, list=18% , signal= 34 %
GO:0003012 tags=43% , list= 19 %, signal=36%
core_enrichment
GO: 0034502 H4C9/ H4C13/ H4C2/ H1-5 / H4C4/ BRCA2/ CDK1/ ESCO2/ H4C3/ TTK/ KNL1/ H4C1/ H2AC4/ CENPA/ BUB1B/ HASPIN/ AURKB/ LEF1/ H4C5/ PLK1/ MTBP/ ZWILCH/ EZH2/ MMS22L/ H4C12/ H4C11/ MIS18A/ H2AX/ GNL3L/ MSH2/ VRK1/ RCC2/ TONSL/ SPDL1/ TERT/ CCT6A/ CENPQ/ RUVBL2/ MACROH2A1/ RHNO1/ KNTC1/ GNL3/ XRCC4/ RB1
GO: 0006261 MCIDAS/ EXO1/ MCM10/ BRCA2/ GINS1/ CDC6/ DACH1/ MCM2/ GINS2/ CDC45/ E2F7/ GINS4/ HMGA1/ BLM/ ORC6/ CDT1/ MCM4/ ORC1/ RFC3/ TK1/ RAD51/ WDHD1/ DSCC1/ E2F8/ POLQ/ POLE2/ DBF4/ CCNE2/ PCNA/ FEN1/ BRCA1/ EME1/ MMS22L/ MCM6/ HELB/ FBXO5/ TICRR/ CCNE1/ TRAIP/ ATAD5/ RFWD3/ TIMELESS/ DNA2/ TIPIN/ RECQL4/ MCM5/ POLD2/ MCM3/ TONSL/ TWNK/ POLB/ RRM1/ MCM7/ CAMSAP3/ SSBP1/ POLA1/ RFC2/ PRIM2/ PRIM1/ RFC4/ RFC5/ ZRANB3/ MGME1/ RBBP8/ LIG1
GO: 0006260 MCIDAS/ EREG/ EXO1/ FAM111B/ DTL/ MCM10/ BRCA2/ GINS1/ CDK1/ ESCO2/ CDC6/ DACH1/ MCM2/ GINS2/ CDC45/ E2F7/ GINS4/ HMGA1/ BLM/ ORC6/ CDT1/ CDC25A/ MCM4/ CHEK1/ ORC1/ RFC3/ TK1/ RMI2/ CCNA2/ RAD51/ WDHD1/ DSCC1/ E2F8/ POLQ/ POLE2/ DBF4/ CCNE2/ PCNA/ FEN1/ BRCA1/ EME1/ CHAF1B/ S100A11/ MMS22L/ MCM6/ HELB/ TOP1MT/ FBXO5/ TICRR/ CCNE1/ RPA3/ TRAIP/ ATAD5/ RMI1/ RFWD3/ RUVBL1/ PCLAF/ TP53/ TIMELESS/ DNA2/ CHAF1A/ TIPIN/ RECQL4/ MCM5/ POLD2/ MCM3/ TONSL/ TWNK/ POLB/ RRM1/ RUVBL2/ DNAJC2/ MCM7/ CAMSAP3/ SSBP1/ POLA1/ RFC2/ PRIM2/ PRIM1/ RFC4/ RFC5/ ZRANB3/ ACTL6A/ MGME1/ RBBP8/ LIG1/ NPM2/ USP37/ JADE3/ SET/ ZNF365/ OBI1/ GTPBP4/ ATR/ CDC7/ TOPBP1
GO: 0006936 NR4A1/ CHRNB2/ TBXA2R/ GRIP2/ SSTR2/ SNTA1/ MYL6/ KCNE4/ FGF13/ SRF/ SGCD/ LARGE1/ GAMT/ SCN5A/ PDE4D/ SLMAP/ ADRA1B/ GUCY1A1/ TBX2/ RNF207/ KCND3/ SLC8A3/ SSPN/ ATP8A2/ PRKG1/ RYR2/ RANGRF/ SPX/ SCN3A/ KCNIP2/ TCAP/ KCNJ8/ KIT/ SCN1A/ PRKD1/ PDE5A/ TRPV1/ HTR2B/ SCN1B/ SCN4B/ STAC/ ADRB2/ SLC8A1/ GSN/ MYH13/ TMOD2/ ABCC9/ FGF12/ PABPN1/ KCNIP1/ TRDN/ MYL3/ CHRNB4/ TACR1/ TPCN2/ RYR1/ KCNH2/ P2RX6/ ACTN3/ CAV1/ CHRNA3/ MYH1/ CACNB2/ CACNA1G/ RGS2/ GPER1/ PTGER3/ TPM1/ KLHL41/ ACTA2/ REM1/ CACNA2D1/ CALD1/ SMPX/ SCN3B/ HRC/ SCN11A/ SCN9A/ DMD/ P2RX1/ APBB1/ ATP2B4/ CASQ1/ STAC2/ MYH3/ TRIM63/ ACTA1/ MYOT/ DTNA/ GJC1/ ADRA2B/ NPPA/ DMPK/ SMTN/ KCNA1/ ANK2/ FLNA/ SCN4A/ SCN2B/ TNNT3/ PPP1R12B/ SCNN1B/ RYR3/ MYLK/ PLN/ TMOD1/ ADRA1A/ CTNNA3/ CACNA1C/ KCNA5/ KCNMA1/ GDNF/ TPM2/ MYL1/ MYOCD/ CRYAB/ MYH2/ GSTM2/ MYH7/ MYL9/ CACNA1H/ MYH6/ NOS1/ P2RX2/ SGCA/ MYL2/ GALR2/ GNAO1/ LMOD1/ CASQ2/ CNN1/ SCN7A/ TACR2/ CHRM2/ MYH11/ ATP1A2/ SYNM/ TNNI3K/ DES/ FXYD1
GO: 0001906 CXCL11/ PPBP/ CXCL8/ RAET1L/ SEMG1/ CCL20/ CXCL10/ DEFA6/ CD1A/ GZMB/ PF4/ CXCL3/ LYZ/ CXCL1/ CXCL9/ SERPINB4/ NOS2/ TREM1/ ULBP2/ RNASE7/ ULBP3/ CCL22/ CCL18/ ULBP1/ FCGR3B/ CD1B/ CCL17/ DEFA5/ IL23A/ IFNG/ GZMH/ CXCL6/ CXCL13/ IL23R/ FCGR3A/ FCGR1A/ HLA- DRA/ KRT6A/ GNLY/ GBP5/ CD55/ FCGR2A/ SYK/ NKG7/ CYRIB/ PRF1/ CXCL2/ HPRT1/ LTF/ GZMA/ CRTAM/ HLA- G/ HLA- DRB1/ CCL25/ HAMP/ ROMO1/ CTSH/ CD2/ GAPDH/ APOL1/ CTSC/ CD1E/ IL12RB1/ SH2D1A/ HLA- B/ MICB/ HSPA8/ RASGRP1/ CCL19/ PTPRC/ F2RL1/ HLA- A/ HAVCR2/ GBP3/ RPS19/ CORO1A/ CX3CR1/ PTPN6/ TUBB4B/ H2BC12/ CXCL14/ PVR/ NECTIN2/ CD1C
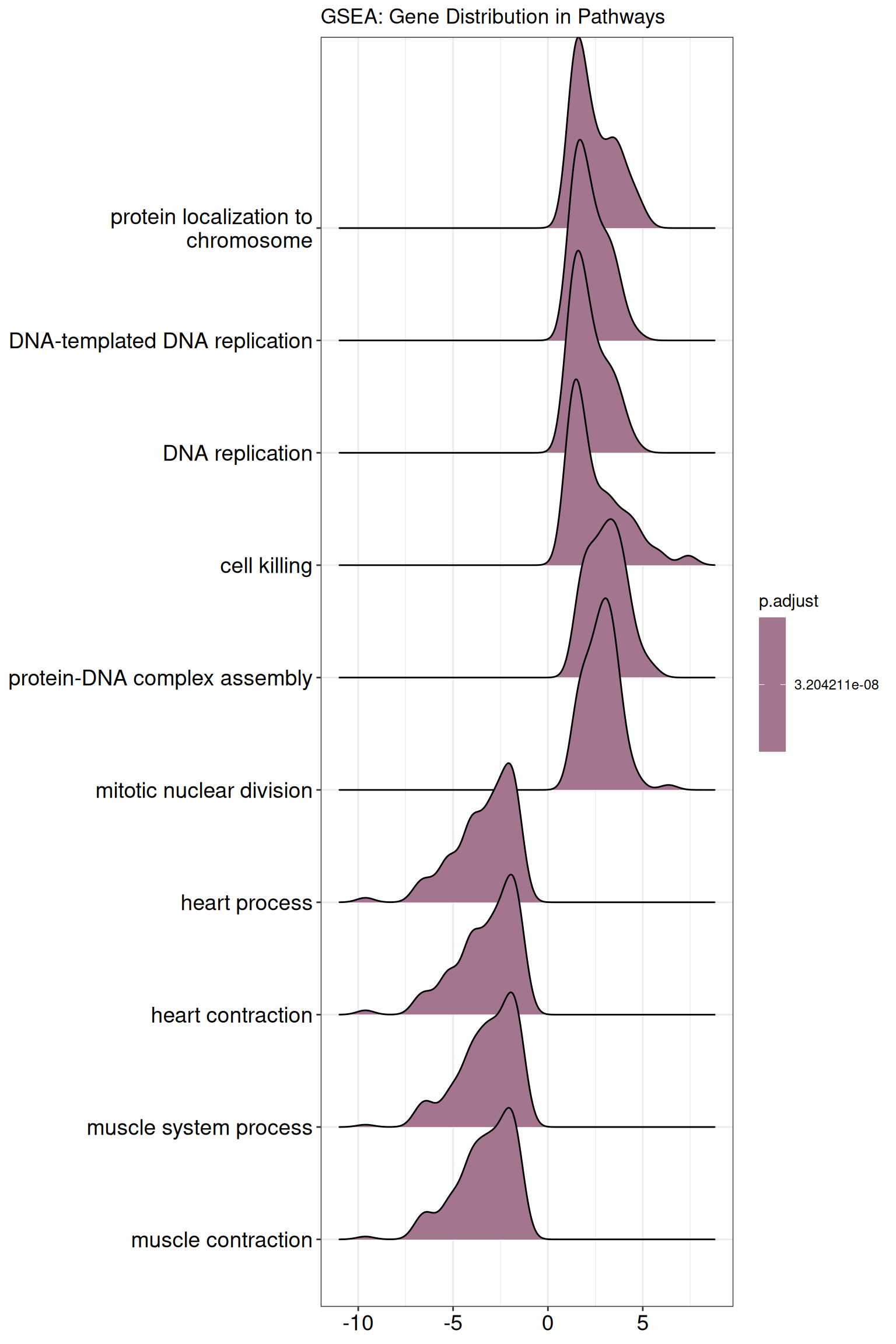
GO: 0003012 TIAM1/ CACNB1/ CLIC2/ VEGFB/ EDN3/ HEY2/ NR4A1/ CHRNB2/ TBXA2R/ GRIP2/ SSTR2/ CFLAR/ SNTA1/ MYL6/ KCNE4/ FGF13/ GATM/ SRF/ IGFBP5/ SGCD/ LARGE1/ GAMT/ CAMTA2/ SCN5A/ NFATC1/ PDE4D/ SLMAP/ ADRA1B/ GUCY1A1/ TBX2/ RNF207/ KCND3/ SLC8A3/ SSPN/ ATP8A2/ PRKG1/ MYOZ1/ RYR2/ RANGRF/ NR4A3/ SPX/ P2RY1/ SCN3A/ KCNIP2/ SLN/ TCAP/ KCNJ8/ KIT/ SCN1A/ PRKD1/ PDE5A/ MSTN/ TRPV1/ HTR2B/ SCN1B/ SCN4B/ STAC/ ADRB2/ SLC8A1/ GSN/ MYH13/ TMOD2/ CAMK2G/ ABCC9/ FGF12/ PABPN1/ KCNIP1/ TRDN/ MYL3/ CHRNB4/ TACR1/ TPCN2/ HDAC4/ RYR1/ KCNH2/ CAMK2B/ P2RX6/ ACTN3/ CAV1/ CHRNA3/ MYH1/ CACNB2/ PDE9A/ CACNA1G/ RGS2/ GPER1/ PTGER3/ TPM1/ KLHL41/ AKAP6/ ACTA2/ REM1/ COL6A1/ CACNA2D1/ CALD1/ SMPX/ ACACB/ SCN3B/ ABCC8/ HRC/ SCN11A/ SORBS2/ SCN9A/ DMD/ P2RX1/ APBB1/ ATP2B4/ CASQ1/ STAC2/ MYH3/ TRIM63/ ACTA1/ MYOT/ FBXO32/ DTNA/ GJC1/ ADRA2B/ NPPA/ PI16/ DMPK/ SMTN/ KCNA1/ ANK2/ FLNA/ SCN4A/ KLF15/ SCN2B/ TNNT3/ PPP1R12B/ SCNN1B/ RYR3/ MYLK/ PLN/ TMOD1/ ADRA1A/ CTNNA3/ CACNA1C/ KCNA5/ MYOZ2/ KCNMA1/ GDNF/ TPM2/ MYL1/ MEIS1/ MYOCD/ CRYAB/ MYH2/ GSTM2/ ASB2/ MYH7/ MYL9/ CACNA1H/ MYH6/ NOS1/ P2RX2/ SGCA/ MYL2/ GALR2/ GNAO1/ LMOD1/ CASQ2/ CNN1/ MYOC/ SCN7A/ TACR2/ CHRM2/ MYH11/ ATP1A2/ HAND2/ SYNM/ TNNI3K/ DES/ FXYD1 結果を可視化します。 dotplotとridgeplotを表示します。 ridge plotとは
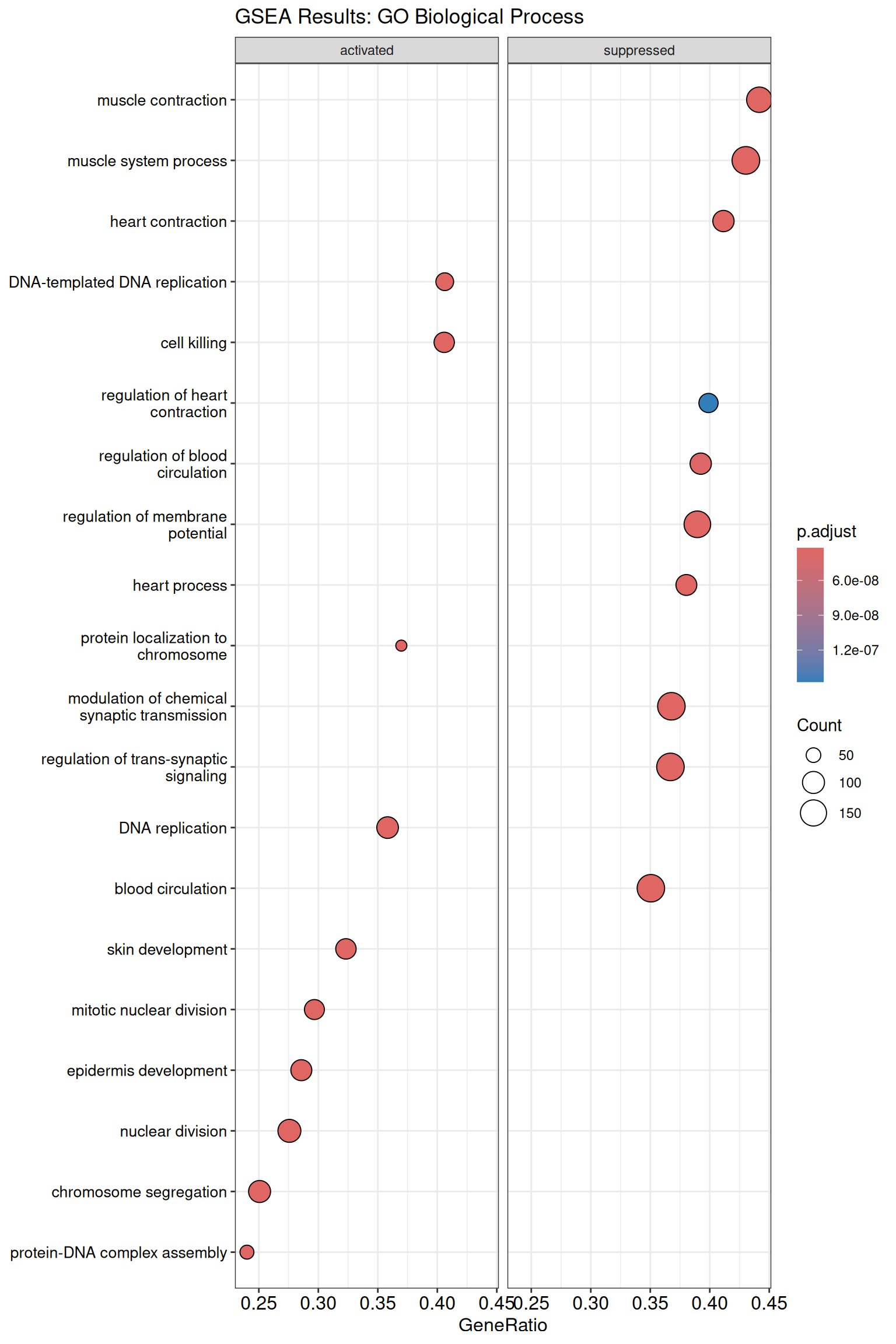
# 横軸はlogFCの分布を示しています。 右に山が寄っていればlogFCが大きい遺伝子が多数含まれているということです。 それぞれのパスウェイの遺伝子の分布パターンを直感的に知ることができます。 dotplotで可視化 # GSEA結果の可視化
# NES(Normalized Enrichment Score):正→upregulated、負→downregulated
dotplot (gse, showCategory = 10 , split = ".sign" ) +
facet_grid (.~.sign) +
labs (title = "GSEA Results: GO Biological Process" ) +
theme (axis.text.y = element_text (size = 10 )) ridgeplotで可視化 ridgeplot (gse, showCategory = 10 ) +
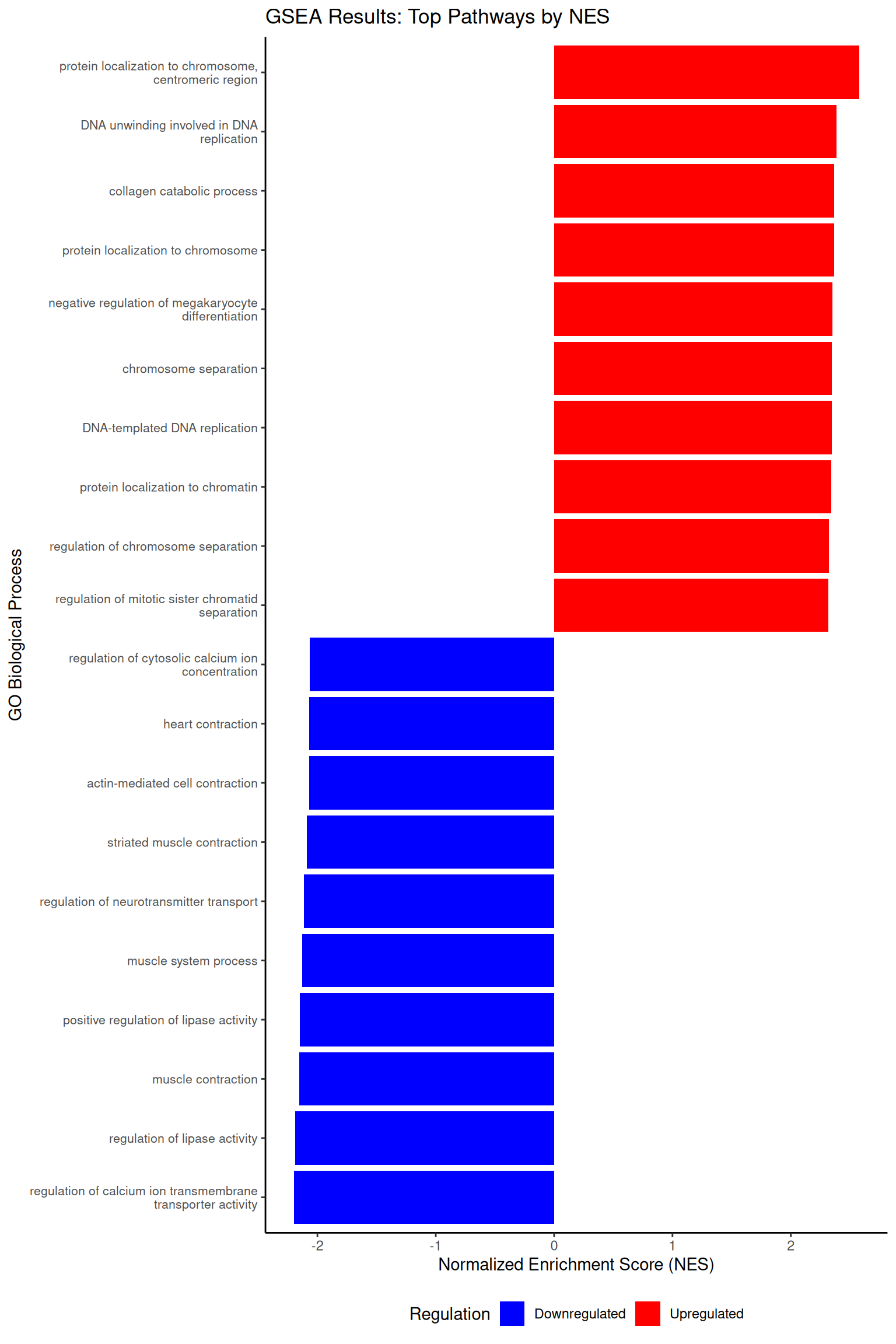
labs (title = "GSEA: Gene Distribution in Pathways" ) Picking joint bandwidth of 0.457 次にbarplotで表示してみます。 GSEAの結果はbarplot()関数では表示できないのでggplot2を使って作成します。 barplotで可視化(ggplot2を使用)。NESの上位10個と下位10個を表示 # GSEAの結果を手動でbarplotとして作成
gse_results <- as.data.frame (gse)
# NESの上位10(最も正の値)と下位10(最も負の値)を取得
top_pathways <- gse_results %>%
arrange (desc (NES)) %>%
slice (c (1 : 10 , (n ()-9 ): n ())) %>%
mutate (
Description = str_wrap (Description, width = 40 ),
regulation = ifelse (NES > 0 , "Upregulated" , "Downregulated" )
)
ggplot (top_pathways, aes (x = reorder (Description, NES), y = NES, fill = regulation)) +
geom_col () +
scale_fill_manual (values = c ("Upregulated" = "red" , "Downregulated" = "blue" )) +
coord_flip () +
labs (
title = "GSEA Results: Top Pathways by NES" ,
x = "GO Biological Process" ,
y = "Normalized Enrichment Score (NES)" ,
fill = "Regulation"
) +
theme_classic () +
theme (
axis.text.y = element_text (size = 8 ),
legend.position = "bottom"
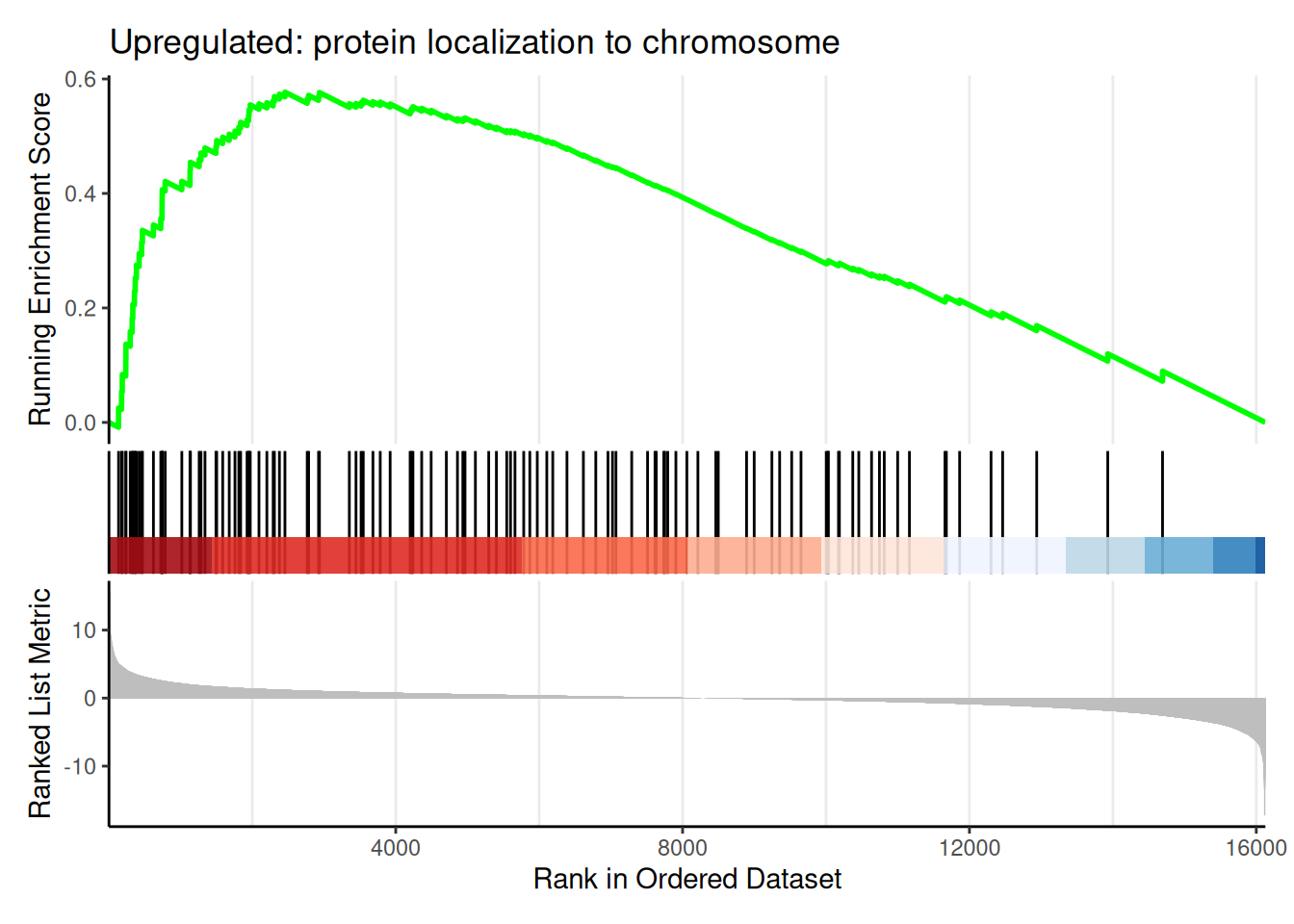
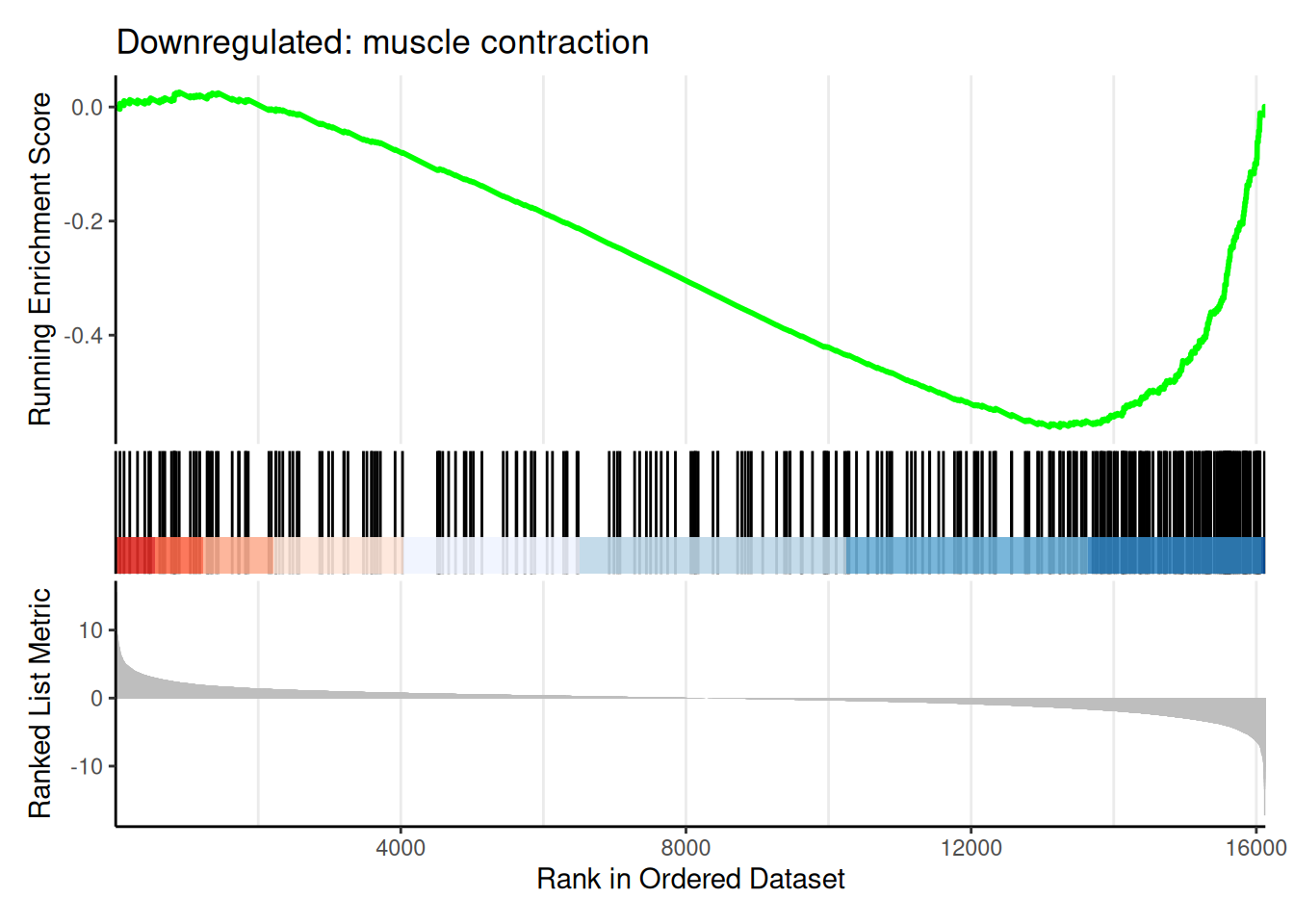
) GSEAの結果はエンリッチメントプロット で表示することが一般的です。 山の頂点が正の方向かつ左に寄っているほど、up-regulatedに有意になりやすいパスウェイです。 山の頂点が負の方向かつ右に寄っているほど、down-regulatedに有意になりやすいパスウェイです。 試しに最もup-regulatedに有意なパスウェイとdown-regulatedに有意なパスウェイを表示してみます。 エンリッチメントプロットで個別パスウェイを表示(最もNESが高いものと最もNESが低いもの) # エンリッチメントプロットで個別パスウェイを詳細表示
gse_results_for_plot <- as.data.frame (gse)
# 最も有意なupregulated(正のNES)パスウェイを選択
most_sig_up <- gse_results_for_plot %>%
filter (NES > 0 ) %>%
arrange (p.adjust) %>%
slice (1 ) %>%
pull (ID)
# 最も有意なdownregulated(負のNES)パスウェイを選択
most_sig_down <- gse_results_for_plot %>%
filter (NES < 0 ) %>%
arrange (p.adjust) %>%
slice (1 ) %>%
pull (ID)
# upregulatedパスウェイのエンリッチメントプロット
up_pathway_name <- gse_results_for_plot %>%
filter (ID == most_sig_up) %>%
pull (Description)
gseaplot2 (gse, geneSetID = most_sig_up,
title = paste ("Upregulated:" , up_pathway_name))
# downregulatedパスウェイのエンリッチメントプロット
down_pathway_name <- gse_results_for_plot %>%
filter (ID == most_sig_down) %>%
pull (Description)
gseaplot2 (gse, geneSetID = most_sig_down,
title = paste ("Downregulated:" , down_pathway_name)) ORAとGSEAの結果比較(有意なパスウェイ数をそれぞれ表示) # ORAとGSEAの結果比較
print ("=== 結果の比較 ===" )
print (paste ("Upregulated有意パスウェイ数:" , sum (ora_result_up@ result$ p.adjust < 0.05 )))
print (paste ("Downregulated有意パスウェイ数:" , sum (ora_result_down@ result$ p.adjust < 0.05 )))
print (paste ("GSEA有意パスウェイ数:" , sum (gse_results$ p.adjust < 0.05 )))[1] "=== 結果の比較 ==="
[1] "Upregulated有意パスウェイ数: 333"
[1] "Downregulated有意パスウェイ数: 350"
[1] "GSEA有意パスウェイ数: 647"